Přírodovědecká fakulta Masarykovy univerzity
Ing. Martin Krsek, CSc.
Metody studia diverzity založené na extrakci nukleových kyselin
I přes nesporný význam metod stručně zmíněných v předcházející kapitole, nejrozšířenějšími metodami studia diverzity nejrůznějších ekosystémů jsou bezesporu metody založené na studiu genetické informace uložené v R/DNA. K jejich využití je nukleové kyseliny nutné z biologického materiálu nejprve izolovat, separovat a koncentrovat. Poté následuje jejich zpracování a využití v navazujících aplikacích. Podívejme se proto nejprve na metody extrakce nukleových kyselin.
Pro izolaci půdní mikrobiální R/DNA existují dva základní postupy, označované jako přímá (in situ) a nepřímá (ex situ) izolace R/DNA. V případě přímé izolace jsou mikrobiální buňky lyzovány přímo v půdní matrici a uvolněná R/DNA je pak následně extrahována a čištěna. Při nepřímé izolaci jsou nejprve mikrobiální buňky extrahovány z půdy, odděleny od půdní matrice a teprve takto získaná mikrobiální frakce je podrobena lyzi. Protože způsoby lyze mikrobiálních buněk v případě přímé i nepřímé izolace jsou téměř identické, všimněme si nejprve nepřímé izolace, tj. způsobů extrakce půdní mikrobiální frakce.
Extrakce půdní mikrobiální frakce
Půda je komplexní prostředí sestávající z fáze pevné, tekuté a plynné. Částice pevné fáze se shlukují do mikro- a makroagregátů a vytvářejí tak půdní strukturu. Jen dobré strukturní půdy zajistí rostlinám vhodné prostředí pro jejich zdárný vývoj. Významný podíl na vytváření půdní struktury mají mikrobiální buňky, které spolu s kořeny rostlin nejrůznějšími mechanismy poutají drobné minerální částice dohromady (sekrece polysacharidů, glykoproteinů a podobných látek, mechanický účinek bakteriálních fimbrií, myceliárních mikroorganismů – hub a některých aktinobakterií spolu s kořeny rostlin, elektrostatické síly a další) a vytvářejí tak půdní agregáty. Mikrobiální buňky nejsou v rámci struktury půdy univerzálně rozšířené a najdeme je především na vnější straně agregátů a v mikroskopických pórech mezi nimi (jedná se ale o nepatrné procento všech půdních pórů), kde mají zajištěny vhodné podmínky pro svou existenci, především dostatek vody a určité množství živin. Důsledkem jejich soustředění v mikropórech i jejich schopnosti poutat dohromady minerální částice půdy (a tímto způsobem být samy k minerálním částicím poutány) je však také snížená dostupnost mikrobiálních buněk pro většinu lytických postupů a jejich uvolnění z půdní matrice není snadné. Proto většina metod izolace půdní mikrobiální frakce kombinuje mechanické metody rozrušení struktury půdy s chemickým působením pufrů různého složení.
Metody izolace mikrobiální frakce se liší způsoby mechanického ošetření půdy (u publikovaných metod však často chybí technická data použitých zařízení), složením pufru použitým pro homogenizaci vzorku i způsobem extrakce mikrobiální frakce samotné po homogenizaci půdních vzorků. Uveďme si několik příkladů izolace půdní mikrobiální frakce.
Jedněmi z prvních, kdo využili metodu nepřímé izolace půdní mikrobiální DNA, byli v roce 1977 Faegri et al., kteří homogenizovali půdu ve vhodném pufru a kombinací nízkootáčkové a vysokootáčkové centrifugace (diferenciální centrifugace) oddělili od sebe frakci obsahující většinu půdních částic spolu s houbovým myceliem a částí bakterií (sediment po první nízkootáčkové centrifugaci) a frakci obsahující 50–80 % všech přítomných bakterií (sediment po vysokootáčkové centrifugaci supernatantu z první nízkootáčkové centrifugace).
Bakken (1985) testoval tři druhy homogenizátorů: Waring blender (obdoba kuchyňského mixéru), „Braun melsungen cell homogenizer“ (třepání půdy se skleněnými kuličkami) a Ilado X 10/20homogenizer (intenzivní mixování vzorku), kde jako homogenizační medium použil vodu, detergenty nebo pufry: 0,22% hexametafosfát sodný pH 8,5 s Na2CO3; 0,3% pyrofosfát sodný; roztok Winogradského; 0,2% bromhexin chlorid; 0,5% Tween 80. Kromě toho testoval možnost flokulace jílových minerálů s použitím okyselení půdního homogenátu na pH 3 (přidáním kyseliny octové a sírové) a přídavkem CaCl2. Půdní homogenát centrifugoval 15 minut ve výkyvném rotoru při 630–1060xg a vzniklý sediment podrobil následně opakované extrakci. Výsledný kombinovaný supernatant centrifugoval 20 minut při 10.000xg. Mikrobiální frakci ze vzniklého sedimentu pak získal pomocí centrifugace v hustotním gradientu za použití Ludoxu HS 40 (Du Pont Co., Wilmington, Del.), případně Percollu (Pharmacia) – 1,16 g/ml.
Macdonald (1986a,b,c) publikoval v roce 1986 sérii tří článků zabývající se izolací půdní mikrobiální frakce. Půda byla 2 hodiny třepána se skleněnými kuličkami v přítomnosti sodné formy iontoměniče Dowex A1 (Sigma) v 0,1% roztoku cholátu sodného a poté následovalo vyplavování bakteriální frakce. Pro disperzi polymerů poutajících mikrobiální buňky k půdní matrici byly testovány různá média: voda, Tween 80 (1% v/v), Triton X-100 (0,1% v/v), N-lauroyl sarcosinát sodný (0,1% w/v) a cholát sodný (0,1% w/v), kde nejúčinnější se jevil cholát sodný (Tab. 2). Dalším testovaným ošetřením byla centrifugace při 1xg, 20xg a 1600xg. Získaná bakteriální frakce byla podrobena centrifugaci v Percollovém gradientu (1,139–1,101 g cm−3).
| Treatment | Yield %a | |
| Water | 26 | |
| Tween 80 | (1% v/v) | 76 |
| Triton X-100 | (0,1% v/v) | 57 |
| Na-N-lauroyl sarcosinate | (0,1% w/v) | 37 |
| Na-cholate | (0,1% w/v) | 84 |
| Výnos vypočten procenty váhy biomasy určené fumigací | ||
(Převzato z: Macdonald, 1986a)
Steffan et al. (1988) získal bakteriální frakci homogenizací půdy v 0,1M fosfátovém pufru pH4,5 s přídavkem polyvinylpolypyrolidonu (PVPP) za použití Waring blendru (mixéru). Po třech jednominutových intervalech mixování přidal do suspenze SDS a následovalo krátké (5 sec) mixování. Poté byly vzorky po dobu jedné minuty třepány v ruce a centrifugovány 10 minut při 1000xg. Vzniklý sediment byl podroben dalším dvěma cyklům popsané extrakce a kombinovaný supernatant centrifugován 30 minut při 10.000xg. Pro snížení obsahu organického materiálu nebakteriálního původu byl sediment 2× resuspendován v 0,1% roztoku hexametafosfátu a pyrofosfátu sodného a poté centrifugován (10.000xg). Finální promytí k odstranění huminových nečistot bylo provedeno v pufru podle Crombacha (0,33 M Tris, 0,001M EDTA).
Hopkins et al. (1991a) testoval devět různých metod půdní disperze, většinu z nichž nakonec zahrnul do následující metody: půda byla mixována v 0,1% roztoku cholátu sodného, poté byl přidán další cholát sodný spolu se sodným chelatačním činidlem (sodium chelating resin) a skleněnými kuličkami a suspenze byla 2 hodiny třepána. Následovala dvouminutová centrifugace při 500xg, poté byl sediment resuspendován v Tris pufru a opět po dobu 1 hodiny třepán, znovu centrifugován (500xg), resuspendován v cholátu sodném, lehce sonikován ve vodní lázni a s přídavkem dalšího cholátu sodného třepán po dobu jedné hodiny. Následovala centrifugace (500xg), resuspendování v Tris pufru, centrifugace (500xg), resuspedování v destilované vodě s následným třepáním (1 hod), centrifugace (500xg), resuspendování v destilované vodě, třepání (1 hod) a poslední centrifugace (500xg).
V následující publikaci pouzili Hopkins et al. (1991b) první krok výše uvedené metody: půda byla mixována v 0,1% roztoku cholátu sodného, poté byl přidán další cholát sodný spolu se sodným chelatačním činidlem (sodium chelating resin) a skleněnými kuličkami a suspenze byla 2 hodiny třepána. Bakteriální frakce byla poté vyplavována proudem fyziologického roztoku.
Holben (1994) použil k homogenizaci roztok Winogradského s přídavkem askorbátu sodného a okyseleného PVPP. Vzorek mixoval 3×1 minutu (Waring blender), poté jej podrobil centrifugaci (640xg) a následně proces homogenizace sedimentu opakoval dvakrát. Bakteriální frakci ze všech tří získaných supernatantů oddělil centrifugací při 14.740xg.
Lindal a Bakken (1995) testovali několik metod fyzikální a chemické disperze půdy. Z fyzikálních metod to byla ultrasonikace za použití sondy o průměru 13 mm a dvou hladinách energie (40W a 55W), mixovaní (Waring blender), použití gumového rotujícího pístu ve zkumavce o průměru o 0,5–1 mm větším než píst a dvouhodinové třepání. Pro chemickou disperzi byl testován roztok deoxycholátu sodného s polyetylén glykolem 6000 a iontoměničem Chelex 100. Účinnost disperze byla hodnocena nízkorychlostní centrifugací (1000xg) nebo centrifugací v Nycodenzovém gradientu.
Courtois at al. (2001) homogenizoval půdu mixováním v 0,9% roztoku NaCl (Waring blender) a z takto získané suspenze oddělil mikrobiální frakci vysokorychlostní centrifugací na Nycodenzovém gradientu (1,3 g ml−1).
Bylo by možné dál pokračovat ve výčtu publikovaných metod izolace půdní mikrobiální frakce, nicméně jak je zřejmé z již uvedeného přehledu vybraných publikací, největší variabilitu vykazuje složení pufru použitého k homogenizaci půdy. Odhlédneme-li od jeho složení, je možné metody rozdělit do dvou skupin podle způsobu vlastní extrakce mikrobiálních buněk na metody používající diferenciální centrifugaci (Faegri et al., 1977, Steffan et al., 1988, Hopkins et al., 1991a,b, Holben 1994) a metody využívající centrifugaci v hustotním gradientu (Bakken 1985, Macdonald 1986a,b, Lindal a Bakken 1995, Courtois et al., 2001).
Účinnější separace mikrobiální frakce od půdních koloidů a především huminových látek je možné dosáhnout centrifugací v hustotním gradientu. Princip této metody je založen na skutečnosti, že bakterie mají menší vznášivou hustotu než většina půdních částic. Jedním z prvních, kdo tuto metodu v kombinaci s nízkootáčkovou centrifugací použil, byl Bakken (1985), který připravil hustotní gradient za použití Ludoxu HS 40 (Du Pont Co., Wilmington, Del) a později velmi často používaného Percollu (Pharmacia Fine Chemicals, Uppsala, Sweden). Percoll tvoří suspenze koloidního křemíku, která má v požadovaném rozsahu hustoty nízkou viskozitu a osmolaritu. Problémem mohou ale být právě částice křemíku zvláště při následném použití fluorescenční mikroskopie díky silné fluorescenci těchto částic. V poslední době získal na popularitě Nycodenz, neionický iodovaný derivát benzoové kyseliny se třemi bočními alifatickými hydrofilními řetězci. Jeho systemický název je 5-(N-2,3-dihydroxypropylacetamido)-2,4,6-tri-iodo-N,N‘-bis(2,3dihydroxypropyl) isophthalamid, molekulová hmotnost 821, hustota 2,1 g ml−1. Není toxický a je velmi dobře rozpustný ve vodě. Jeho vlastnosti poprvé popsali Rickwood et al. (1982) a pro účely izolace půdní mikrobiální frakce ho jako jedni z prvních použili Lindahl a Bakken (1995). Z poslední doby můžeme uvést například práci Liu et al. (2010), kteří Nycodenzový gradient použili pro izolaci bakterií z půdy při přípravě metagenomové BAC knihovny. Práci s Nycodenzovým gradientem popíšeme podrobněji v následujících kapitolách práce.
Konečně bychom také měli zmínit metodu využívající zachycení cílové skupiny mikroorganismů na magnetických nosičích a jejich následné oddělení od půdní suspenze. V tomto případě jsou magnetické mikroskopické kuličky obalené monoklonálními či polyklonálními protilátkami smíchány s půdním homogenátem, kde se na ně nachytají cílové mikroorganismy, které jsou potom pomocí magnetu odděleny od zbytku půdního homogenátu. Metoda detekce určitých skupin mikroorganismů pomocí protilátek se většinou využívala především pro detekci lékařsky významných mikroorganismů ve vodě a potravinách, ale její aplikovatelnost pro půdní podmínky potvrdila již v roce 1968 studie Schmidta et al., kteří využili techniku fluorescenčních protilátek ke studiu volně žijících bakterií, jako jsou rhizobia. Přímo metodu imunomagnetické detekce pro spory bakterií Streptomyces lividans v půdním prostředí využili například Wipat et al. (1994).
Extrakce R/DNA z půdy a půdní mikrobiální frakce
Extrakci půdní mikrobiální R/DNA se věnuje celá řada článků, což je zřejmě způsobeno především velkou variabilitou půdních podmínek znemožňujících vytvoření jedné univerzální metody extrakce R/DNA. Složitost extrakce se také odráží i v množství komerčních kitů k izolaci nukleových kyselin z půdy a sedimentů, o kterých se zmíníme v závěru této kapitoly. Na tomto místě se budeme věnovat „klasickým“ postupům izolace/extrakce nukleových kyselin, které i z hlediska výuky a přípravy absolventů vysokých škol považujeme za mnohem vhodnější (ve srovnání s používáním komerčních kitů). Celý proces extrakce je možné rozdělit do několika fází, kde se každá podílí na kvalitě konečného produktu: volba vhodného pufru, lyze buněk a konečně čištění uvolněné R/DNA.
Volba vhodného pufru
Prvním krokem je volba vhodného pufru, ve kterém se uskuteční vlastní lyze mikrobiálních buněk. Jde o krok zdánlivě méně důležitý, který však významnou měrou rozhoduje o výsledku celé extrakce. Úlohou pufru je zajistit vhodné prostředí pro lyzi buněk (například často využívaný lysozym je inhibován vysokou koncentrací solí v roztoku), zabránit degradaci R/DNA uvolněné lyzí (především působením restrikčních enzymů) a v případě přímé extrakce R/DNA také zabránit vazbě uvolněných nukleových kyselin na půdní koloidy. A právě zde dochází ke „střetu zájmů“, protože z hlediska izolace a ochrany R/DNA po jejím uvolnění je žádoucí mírně alkalické pH (většinou pH 8) a vysoká chelatační schopnost pufru (restrikční enzymy potřebují bivalentní ionty), tedy podmínky vhodné i k uvolnění kontaminujících huminových látek. Jakmile však dojde k uvolnění těchto látek, jsou při většině extrakčních procesů vysráženy spolu s R/DNA a jen velmi obtížně se z roztoku odstraňují.
Za tradiční pufr bychom mohli označit fosfátový pufr (například 0,12 M fosfátový pufr pH 8), který umožňuje izolaci značného množství R/DNA, bohužel však současně uvolňuje mnoho huminových látek. Tento pufr byl použit v mnoha pracích, namátkou jmenujme alespoň některé z nich: Ogram et al. 1987, Leff et al. (1995), Stefan et al. (1988). Další velkou skupinu tvoří pufry s různým obsahem EDTA a TrisHCl; EDTA vázáním bivalentních iontů zabraňuje činnosti restrikčních enzymů a TrisHCl zajišťuje vhodné pH roztoku: jmenujme například pufr použitý Crombachem et al. (1972) a dále využívaný Torsvik et al. (1990a) obsahující 33mM Tris-HCl a 1 mM EDTA, pH 8,0. Picard et al. (1992) použil TNPE pufr: 50mM Tris-HCl, 10 mM EDTA, pH 8,0, 100 mM NaCl, 1% (w/w) polyvinylpolypyrolidone (PVPP); tento pufr později použili také například Frostegard et al. (1999), Courtois et al. (2001) a další; EDTA najdeme ve většině dalších pufrů použitých k izolaci půdní mikrobiální DNA (např. Torsvik et al., 1980; Liles et al., 2008).
Lyze mikrobiálních buněk
Metody používané k lyzi mikrobiálních buněk je možné rozdělit na dvě skupiny, na metody mechanické a chemicko-enzymatické. Většina postupů extrakce R/DNA používá kombinaci obou přístupů, kdy především při přímé lyzi buněk v půdní matrici mechanické ošetření v počáteční fázi extrakce zajišťuje kromě přímého lytického účinku i přístup použitých enzymů a chemikálií k mikrobiálním buňkám pevně vázaným v matrici půdy.
Nejběžnějším mechanickým ošetřením je intenzivní třepání půdní suspenze často s přidáním různě velkých skleněných kuliček. Například Hopkins et al. (1991a,b) nebo Macdonald (1986a) použili třepání půdní suspenze se skleněnými kuličkami především k dokonalému resuspendování či homogenizaci vzorku za účelem zpřístupnění mikrobiálních buněk lytickému účinku použitých enzymů a ostatních sloučenin. Velmi účinnou variantou tohoto ošetření je tzv. beat beating, tj. velice intenzivní třepání půdy (4–6,5 m/s) s pufrem a skleněnými kuličkami o velikosti kolem 0,1 mm, kde už kromě homogenizace vzorku dochází i k mechanické lyzi části mikrobiálních buněk. Původně šlo o velmi hmotné a hlučné zařízení (Braun Beat –beater), kdy vzorek půdy (několik gramů dle velikosti použitých zkumavek) s příslušným pufrem a skleněnými kuličkami (0,11 mm) 5 minut rotoval ve skleněné zkumavce ve speciální kapsuli přístroje chlazené stlačeným CO2 (Smalla et al., 1993, van Elsas et al., 1997). Toto zařízení bylo v posledních letech narazeno menšími přístroji (nazývanými např. FastPrep, Ribolyzer, MagnaLyser a podobně), kde je rotační pohyb jedné zkumavky nahrazen vertikálním třepáním 12, případně 24 dvoumililitrových plastových zkumavek se vzorkem (např. 0,5 g půdy) a příslušnou lytickou matricí obsahující kromě jiného i různě velké kuličky. Nejznámější je systém FastPrep, což je označení jak pro samotný homogenizátor, tak i pro komerčně dodávané a velmi úspěšné kity pro izolaci R/DNA z půdy, bakterií, mikromycet a podobně (Griffiths et al., 2000; Dineen et al., 2010a, 2010b; Mettel et al., 2010).
Další možností je sonikace vzorku budˇve vodní lázni, nebo podstatně účinnější způsob za použití kovové sonikační sondy vibrující v nádobce s půdní suspenzí nebo mikrobiální frakcí. Zvláště při použití sondy se dosáhne velmi účinné lyze buněk, bohužel však spojené s intenzivním nastříháním extrahované DNA a proto se tento způsob příliš neuplatňuje (Frostegard et al., 1999; Courtois et al., 2001).
Z dalších možností musíme jmenovat tradiční „freezing-thawing“, tedy prudké zmrazování a rozmrazování půdní suspenze za použití tekutého dusíku a teplé vodní lázně. Avšak vzhledem k malé velikosti především bakteriálních buněk a velké rychlosti jejich zmrazení se dá předpokládat, že toto ošetření nebude na značnou část půdních bakteriálních buněk příliš účinné. Smalla et al. (1993) porovnávala účinek tohoto ošetření s tradičním beat-beatingem, který prokázal podstatně vyšší účinnost lyze a Kauffmann et al. (2004) a Zhou et al. (1996) došli k závěru, že toto ošetření je účinné pouze na Gram-negativní buňky.
Byly také testovány nejrůznější způsoby tření či mletí půdy, a to půdy suché (Frostegard et al., 1999), nebo zmražené tekutým dusíkem (Hurt et al., 2001). Frostegard et al. (1999) a Courtois et al. (2001) využili strojové mletí v achátové třecí misce s achátovými kuličkami o průměru 20 mm po dobu až 60 minut nebo drcení wolframovými kroužky po dobu 30 vteřin, avšak i po tomto ošetření zůstalo podle mikroskopického pozorování více než 50 % buněk intaktních. Tření půdy s kapalným dusíkem ručně v třecí misce použili k extrakci DNA z půdy Volossiouk et al. (1995). Zhou et al. (1996) testoval účinek tohoto ošetření na kultury Gram-pozitivních buněk smíchaných se sterilním pískem, kde toto ošetření zvýšilo výnos DNA 2–6× ve srovnání s pouhým freezing-thawing.
Nejčastěji používaným enzymatickým ošetřením je působení lysozymu. Tento enzym přítomný ve slinách, slzách a jiných tělních exkretech, ale také ve vaječném bílku, hydrolyzuje 1–4 beta glykosidickou vazbu mezi N-acetylmuramovou kyselinou a N-acetyl-D-glukozaminem v peptidoglykanu bakteriální buněčné stěny především Gram-pozitivních bakterií a mezi N-acetyl-D-glukózaminovými zbytky v chitodextrinu mikromycet (zřejmě proto se někdy lysozym používá pro izolaci R/DNA z mikromycet). Gram-negativní bakterie vzhledem k odlišnému složení buněčné stěny jsou chráněny proti ataku lysozymu, avšak tato ochrana není úplná (Masschalck et al., 2001). Lysozym je inhibován vysokou koncentrací solí, což je nutné respektovat při výběru pufru použitého k lyzi. K pufru bývá někdy přidávána sacharóza, která stabilizuje sféroplasty vznikající účinkem lysozymu do přidání detergentu lyzujícího cytoplazmatickou membránu. Kromě lysozymu (a často společně s ním) bývá také užívána proteináza K, širokospektrá serin-proteáza používaná k trávení proteinů a odstraňování kontaminací při extrakci nukleových kyselin. Její lytickou činností jsou také rychle rozštěpeny endonukleázy a tímto způsobem jsou uvolněné nukleové kyseliny chráněny proti jejich degradaci. Svou proteolytickou schopnost si proteináza K uchovává i za přítomnosti SDS a EDTA, což je nesmírně důležité právě při procesech izolace a čištění R/DNA. Z dalších enzymů můžeme zmínit achromopeptidazu, kterou Ezaki a Suzuki (1982) použili k lyzi Gram-pozitivních anaerobních koků rezistentních účinku lysozymu. Tento enzym atakuje peptidové můstky mezi řetězci peptidoglykanu.
K lyzi mikromycet lze použít kombinaci lytických enzymů z Rhizoctonia solani, vykazujících glukanázovou, proteázovou, pektinázovou a amylázovou aktivitu a lytických enzymů z Trichoderma harzianum s glukanázovou, cellulázovou, proteázovou a chitinázovou aktivitou (Sigma-Aldrich). Novo Nordisk produkoval Novozym 234 (směs chitinolytických enzymů), který se používal k přípravě houbových protoplastů, extrakci DNA z houbových kultur i k extrakci DNA mikromycet z půdy (Porteous a Armstrong, 1991).
K chemické lyzi bakteriálních buněk je nejčastěji využíván laurylsufát sodný (SDS), tenzid využívaný i v mnoha přípravcích osobní hygieny, jako například zubní pasty a podobně. Pro účely lyze se často používá tzv. horká lyze (hot SDS lysis), kdy je k půdní suspenzi přidán SDS do konečné koncentrace 1 % a výsledná suspenze je inkubována při 65–70oC. Lyze buňky proběhne narušením cytoplazmatické membrány. Dalším často využívaným kationtovým detergentem je cetyltrimetylammonium bromide (nebo také hexadecyltrimethylammonium bromid; CTAB). Tento detergent by neměl být použit spolu s SDS nebo fenolem, protože s nimi tvoří nerozpustné sraženiny. Pro zachování rozpustnosti DNA je potřebná vysoká koncentrace monovalentních kationů jako Na+ nebo NH4+, která také zajistí vysrážení polysacharidových (obecně uhlovodíkových) a proteinových komplexů s CTAB, které budou za těchto podmínek naopak nerozpustné. Podobný účinek mají i další detergenty, lze dokonce použít i kterýkoliv běžný prací prostředek (Bahl a Pfenninger, 1996). Metoda izolace DNA pomocí pracích prostředků byla publikována pro nejrůznější typy tkání (lidské tkáně, tkáně plazů, slimáků, tabáku), v naší laboratoři jsem ji aplikovali i na půdní vzorky a i zde byla izolace úspěšná, což není překvapivé, vezmeme-li v úvahu, že většina pracích prostředků obsahuje detergenty, enzymy a chelatační činidla. Problémem zůstává skutečnost, že podobně jako při využití SDS a EDTA je spolu s DNA uvolněno i mnoho huminových substancí, což se projeví tmavým zbarvením vzorku a je samozřejmě spojeno se značnými komplikacemi při dalším čistění a užití extrahované DNA (inhibice enzymatických reakcí).
Čištění – srážení izolované R/DNA
Po uvolnění z buněk je potřebí R/DNA zbavit kontaminací nukleoidových proteinů i dalších zbytků buněčných struktur a – především v případě přímé lyze – i příměsí z půdního prostředí, hlavně huminových substancí, které vykazují výrazný inhibiční vliv na enzymatické reakce a znesnadňují tak další práci s R/DNA. Úroveň kontaminace je posuzována skanováním na UV spektrofotometru, kde fenolické látky (huminové substance) mají maximum absorbance při vlnové délce 230 nm, DNA při 260 nm a proteiny při 280 nm. K výpočtu kontaminace pak slouží poměry absorbance 260/230 pro huminové látky a 260/280 pro proteiny, které by se oba u čisté DNA měly pohybovat v rozmezí 1,8–2,0.
Problémem huminových substancí je fakt, že jsou běžnými srážecími technikami používajícími etanol nebo isopropanol vysráženy z roztoku spolu s R/DNA a dají se proto jen velmi obtížně odstranit. Proto je lépe soustředit veškeré úsilí na minimalizaci těchto kontaminací již v průběhu procesu extrakce R/DNA, tedy volbou vhodného pufru i lytických ošetření, jak bylo diskutováno výše. Kromě toho byly publikovány postupy omezující kontaminaci přídavkem nejrůznějších chemikálií, z nichž asi nejčastěji diskutovaným je již výše zmíněný polyvinylpolypyrolidone (PVPP), běžně používaný jako plnidlo (tablety – např. náhražky cukru) a čeřící látka (nápoje).
PVPP je makromolekulární ve vodě nerozpustná sloučenina, která je schopna na sebe selektivně vázat fenolické látky (používá se např. pro vazbu tříslovin při výrobě piva). Reakce probíhá jenom v určité oblasti pH. V alkalickém prostředí se vazba uvolňuje a adsorbované látky přecházejí zpět do roztoku. Právě tento fakt může působit komplikace při extrakci R/DNA, kde se využívají pufry s alkalickým pH. To je zřejmě také důvod, proč některé metody využívající PVPP při extrakci nukleových kyselin hovoří o tzv. „acid washed PVPP“, tedy okyseleném PVPP. PVPP je součástí například již výše zmíněného TNPE pufru, který byl publikován Picardem et al. (1992) a dále používán Frostegardem et al. (1999), nebo součástí protokolu izolace půdní bakteriální DNA publikované Holbenem (1994). Jiné metody využívají PVPP jako filtrační kolonu (Lakay et al., 2006; Arbeli a Fuentes, 2007).
Podobný účinek jako PVPP má mít již dříve zmíněný CTAB, který by tedy kromě úlohy při narušení cytoplazmatické membrány měl být schopen tvořit nerozpustné komplexy s huminovými látkami a již výše zmíněnými polysacharidy a proteiny (Obr. 3-1).
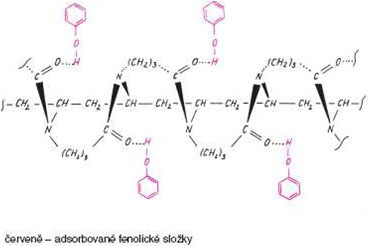
Obr. 3-1 Vazba fenolických sloučenin na CTAB
Kromě PVPP byl testován i vliv polyvinylpyrolidonu (PVP) na čistotu DNA V tomto případě byl rozpustný PVP přidán do agarózového gelu, kde zpomalil elektroforetickou mobilitu fenolických látek (huminové substance) a umožnil tak jejich oddělení od nukleových kyselin (Young et al., 1993).
Další možností odstranění kontaminace huminovými látkami jsou nejrůznější filtrační kolony. Kromě již zmíněného PVPP se používají kolony Sepharozové (Torsvik 1980), Sephadexové (Howeler et al., 2003, Miller et al., 1999), Chelexové (Straub et al., 1994) častěji využívané v prvních fázích izolace k usnadnění lyze buněk a existuje i celá řada komerčně produkovaných filtračních kolon, jako například Wizardové kolony (Henne et al., 1999) nebo Elutip-d kolony (Degrange a Bardin 1995; Frostegard et al., 1999), nebo kolony, které jsou součástí komerčních kitů pro izolaci půdní DNA (např. FastDNA® SPIN Kit for Soil, FastRNA® Pro Soil-Direct Kit, FastRNA® Pro Soil-Indirect Kit). Při použití filtračních kolon dochází buď k vazbě huminových látek na tyto kolony (např. Sepharozové kolony), DNA v tomto případě kolonami volně prochází, nebo zvláště u kolon, které jsou součástí izolačních kitů, se naopak nukleové kyseliny selektivně váží na matrici kolon, zatímco huminové látky jsou vymyty. Po změně pH nebo osmotické síly roztoku jsou čisté nukleové kyseliny uvolněny z kolon.
K získání a čištění R/DNA po lyzi se používají také nejrůznější srážení. Nejčastější je srážení etanolem nebo isopropanol za vysoké koncentrace solí v roztoku (obvykle 0,3 M acetát sodný), kde základním rozdílem je množství alkoholu použitého ke srážení (2–2,5 objemů v případě etanolu, nebo 0,6–1 objem u isopropanolu). V obou případech jsou ale spolu s nukleovými kyselinami vysráženy také huminové látky. Existují však postupy srážení, které jsou selektivnější a při jejich použití dojde ke značnému snížení kontaminace nukleových kyselin těmito látkami. Jedním z nich je srážení R/DNA pomocí polyetylén glykolu (PEG). V tomto případě se většinou používá PEG 6000 nebo PEG 8000 v konečné koncentraci 10 % za přítomnosti 1M NaCl (např. Howeler et al., 2003; Towe et al., 2011). Je možné také využít srážení pomocí 5mM spermine tetrahydrochloride (spermine-HCl), zde je naopak nutná nízká koncentrace solí, tedy nejlépe v TE pufru (Hopwood et al., 1985).
Používají se i další způsoby čištění DNA, jako je centrifugace v gradientu chloridu cesného nebo čištění na hydroxyapatitových kolonách (Ogram et al, 1987, Stefan et al., 1988). V obou případech však jde o procesy jak časově tak finančně náročné, během kterých dochází také ke ztrátám na výnosu DNA. Kromě toho centrifugace v gradientu chloridu cesného je vhodná spíš pro odstranění proteinových a dalších buněčných nečistot, huminové látky budou dispergovány v tomto gradientu (Stefan et al., 1988).
Odstranění dalších kontaminantů, především proteinů, již nepředstavuje takový problém. Část proteinů může být rozložena přídavkem proteinázy K a běžným ošetřením je fenol-chloroformová extrakce. Proteiny mohou být také sráženy acetátem draselným (Smalla et al., 1993) nebo cesium chloridem (Smalla et al., 1993, van Elsas et al., 1997).
Již v úvodu této kapitoly jsme se zmínili o komerčních soupravách/kitech k extrakci DNA z půdy, kterých se zvláště v posledních letech objevila celá řada. Zmiňme například UltraClean Soil DNA kit a PowerSoil™ DNA Isolation Kit (MoBio Laboratoriem Inc., Solana Beach, CA, USA), FastDNA® SPIN Kit for Soil (Q-BIOgene), Mag-Bind® Soil DNA Kit (Omega Bio-Tek, Inc.), nebo ZR Soil Microbe DNA Kit (Zymo Research). S většinou těchto souprav je možné získat velmi kvalitní DNA z většiny půd, avšak podobně jako stále neexistuje univerzální metoda extrakce R/DNA z půdních mikroorganismů aplikovatelná na všechny půdní podmínky (pro úplnost/správnost je zde třeba zmínit metodu izolace DNA z půdy – ISO standard 11063 – Soil quality – Method to directly extract DNA from soil samples), neexistuje také jeden univerzální kit účinný ve všech půdních podmínkách. Kromě toho se v případě komerčních kitů pracuje s „černou skříňkou“, kde se míchají blíže nespecifikované roztoky a používají nejrůznější centrifugační kolony bez podrobnějších informací, s čím se vlastně pracuje, což prakticky znemožňuje jakoukoliv optimalizaci extrakčního procesu. Je také třeba zdůraznit, že i při použití stejných komerčních extrakčních kitů (případně stejných extrakčních protokolů) a stejných půdních vzorků se výsledky izolace budou lišit laboratoř od laboratoře (vlastní nepublikované výsledky; Pan et al., 2010). A konečně z hlediska výuky na vysokých školách nepovažujeme použití kitů za přínosné pro již zmíněnou absenci znalosti procesu samotné extrakce. I přesto je nutné zdůraznit, že pro některé aplikace může být použití kitů vhodné a bylo by chybou se mu a priori vyhýbat.
Metody studia mikrobiální diverzity založené na analýze R/DNA bez použití PCR
Metody založené na hybridizaci DNA izolované z mikrobiální komunity
Jedinou metodou, která je teoreticky schopná určit absolutní diverzitu mikrobiálního společenstva, je metoda reasociace denaturované DNA extrahované z mikrobiálních společenstev. Tuto metodu k analýze diverzity mikrobiálních společenstev poprvé použila v roce 1990a Torsvik et al., ale vzhledem ke své obtížnosti nedoznala velkého rozšíření. Metoda spočívá v extrakci DNA z environmentálního vzorku, jejího naštěpení a tepelné denaturaci a následné renaturaci po ochlazení vzorku, jejíž rychlost je nepřímo úměrná diverzitě analyzovaného vzorku. Čas potřebný k dosažení poloviční reasociace může pak být využit jako index diverzity příslušného vzorku. Jedná se tedy o jednoduchý princip dříve používaný k určení velikosti geonomů nejrůznějších organismů (Baldari a Amaldi, 1976; Dhillon et al., 1980). Jeho aplikace na podmínky mikrobiálních komunit, zvláště půdních, však spolu přináší značné komplikace. Kromě již dříve zmíněného problému zajištění extrakce DNA ze všech mikrobiálních buněk studované mikrobiální komunity společného pro všechny ostatní metody založené na extrakci DNA, bude prvním závažným problémem zajištění DNA v dostatečné kvantitě i kvalitě. Metoda vyžaduje v porovnání s jinými molekulárními metodami značné množství DNA, která musí kromě toho být dostatečně čistá, protože jakékoliv nečistoty (především huminové látky) mají značný vliv na průběh reasociace denaturované DNA. Proto byla extrahovaná DNA v původní metodě (Torsvik et al., 1990a) čištěna na hydroxyapatitových kolonách. Poté byla nastříhána na menší kousky (reasociace intaktní DNA by trvala neúměrně dlouho) a opět čištěna na hydroxyapatitových kolonách z důvodu odstranění malých úseků DNA. Teprve potom mohlo být přistoupeno k tepelné denaturaci DNA a následné reasociaci za nižší teploty. Právě pomalý průběh reasociace (i po nastříhání DNA je k dosažení poloviny reasociace potřeba inkubace v řádu dní až několika týdnů) spolu s dalšími nároky této metody na přístrojové vybavení a podobně zřejmě způsobil, že se tato metoda nerozšířila a naprostá většina publikovaných prací využívající tuto metodu pochází z laboratoře autorky (Torsvik et al., 1990a; Ovreas et al., 2003; Ovreas a Torsvik, 1998; Sandaa et al., 1999; Ritz et al., 1997).
Kromě metody reasociace princip hybridizace DNA používají i další metody, často však již za přispění PCR, či jiných molekulárních metod. Například Griffiths et al. (1996) použili hybridizaci DNA izolované z různých půdních vzorků ke srovnání struktury mikrobiálních společenstev těchto půd. V tomto případě je DNA z jednoho vzorku naštípána restrikčními endonukleázami a označena buď pomocí tzv. „nick translation“, metody, při které je DNA naštípána pomoci DNázy a následně označena s využitím DNA polymerázy I, která zaměňuje některé nukleotidy za značené (Rigby et al., 1977), nebo „random primer labeling“ (Thomas-Cavallin M. a Aït-Ahmed O, 1988), kdy jsou k denaturované DNA přidány náhodné oligonukleotidy, které jsou za pomocí Klenowa fragmentu polymerázy a směsi 4 nukleotidů, z nichž jeden je značený, prodlouženy za produkce značené dvouřetězcové DNA. Takto značená DNA jednoho společenstva je poté hybridizována s DNA druhého vzorku imobilizovaného na membráně. Touto metodou je tedy možné pouze srovnat dva vzorky DNA, ale protože je srovnaní prováděno oboustranně (vzorek A je srovnáván se sondami vytvořenými ze vzorku B a naopak), porovnáním získáme informace jak co do druhové bohatosti, tak i vyrovnanosti obou vzorků.
Konečně jsou na hybridizaci založeny nejrůznější molekulární metody využívající různě specifické DNA sondy a pracující jak s DNA, tak i s RNA, nebo dokonce in situ. Tímto způsobem je možné získat důležité kvantitativní i kvalitativní údaje o studované komunitě. Nutno podotknout, že část těchto metod je opět založená na využití PCR. Sondy mohou mít nejrůznější specificitu od domén až po jednotlivé mikrobiální druhy a mohou být i různě značeny (Theron a Cloete, 2000). Tradičně se používaly radioaktivně značené sondy, v poslední době však nabyly na významu fluorescenční sondy, které vedly k aplikaci techniky FISH (fluorescent in situ hybridization) i v analýze půdních mikrobiálních společenstev (Margesin et al., 2009; Henneberger et al., 2011; Zhang et al., 2012).
Metagenomové knihovny
Konečně je nutné zmínit metodu tvorby metagenomových knihoven, někdy také v literatuře nazývané „shotgun cloning“. Metagenomem rozumíme všechny geny přítomné ve zkoumaném environmentálním vzorku. Metagenomová knihovna tedy vznikne přímým klonováním DNA extrahované z environmentálního vzorku. Genetická informace vytvořených klonů je pak nejrůznějšími způsoby analyzována. Jednu z prvních studií používajících tento přístup publikovali již v roce 1991 Schmidt et al., kteří klonovali DNA izolovanou z mořského bakterioplanktonu do bakteriofága lambda a mezi klony identifikovali a sekvenovali 38 klonů s geny pro 16S rRNA. Dalším mezníkem byla práce Rondona et al. (2000), kteří publikovali jednu z prvních prací zabývajících se tvorbou metagenomových knihoven s použitím bakteriálního umělého chromozómu (bacterial artificial chromosome – BAC) a DNA izolované ze dvou půdních vzorků.
Metagenomové knihovny představují nevyčerpatelný a dosud téměř nedotčený zdroj informací o genetické variabilitě a potenciálu nejrůznějších mikrobiálních společenstev. Praktické provedení tohoto úkolu však není snadné. Základním předpokladem úspěšné tvorby knihovny je totiž klonování velkých úseků DNA, často o velikosti několika stovek kb. Jen takovéto klony vytváří předpoklad k získání opravdu užitečných genetických informací například ve formě nových ucelených metabolických drah a usnadňují následnou analýzu knihoven. Běžnými postupy extrakce mikrobiální DNA jsou však izolovány fragmenty DNA jen o velikosti 10 – 20 kb. K získání větších úseků je většinou potřeba přistoupit k izolaci DNA z mikrobiálních buněk v terčících z nízkotuhnoucí agarózy. Mikrobiální buňky jsou potom v těchto terčících lyzovány a uvolněná DNA naštěpena restrikčními enzymy na fragmenty o vhodné délce. Kontrola účinnosti štěpení DNA spolu se separací DNA podle molekulové hmotnosti fragmentů se provede na pulzní gelové elektroforéze. Z výsledného gelu je pak vyříznuta oblast odpovídající požadované molekulové hmotnosti DNA, která je z gelu získána buď enzymatickým rozpuštěním agarózy, nebo vyplavením pomocí elektroforézy.
Získáním DNA fragmentů o vysoké molekulární hmotnosti však úskalí metody nekončí. Fragmenty je nutné vložit do vhodného vektoru a následně pak analyzovat. Avšak už jen samotná manipulace s takovými fragmenty DNA je velice obtížná – například při pipetování může dojít velice snadno k narušení celistvosti těchto fragmentů (a tedy snížení molekulové hmotnosti získaných fragmentů). Dalším úskalím je výběr vhodného vektoru, který by měl v ideálním případě zajistit neselekční vložení všech získaných DNA fragmentů a případně i jejich expresi ve vhodném organismu. Jako vektor je možné použít plazmidy, častěji jsou ale využívány vektory odvozené od různých fágů, jako například cosmidy, odvozené z fágu lambda. Tento vektor obsahuje cos sekvenci z původního fága (nutnou pro zabalení DNA) a vhodný počátek replikace dle požadavků klonované DNA. V porovnání s normálními plazmidy schopnými pojmout fragmenty 1–20 kb, se cosmidy používají ke klonování fragmentů v rozmezí 37–52 kb. Fosmidy jsou podobné cosmidům, jsou ale odvozené z bakteriálního F-plazmidu. Velikost kolonovaných fragmentů zde dosahuje 40 kb. Vzhledem k nízkému počtu kopií jsou stabilnější než cosmidy s vysokým počtem kopií. Dalším typem klonovacího vektoru jsou tzv. phasmidy (phagemidy). které byly vyvinuty jako hybridy fága M13 a plasmidů. Mohou růst podobně jako plasmidy, zároveň však mohou obsahovat i jednořetězcovou DNA jako virové částice. Významným přínosem ve tvorbě metagenomových knihoven bylo vytvoření výše zmíněného BAC vektoru založeného na F-plazmidu. Na rozdíl od fosmidů jsou schopné pojmout až 150–350 kb genetické informace. Z bakteriálního P1-plasmidu byl vyvinut podobný vektor nazývaný PAC. Jen pro úplnost zde zmiňme vektory vyvinuté pro klonování DNA z jiných než bakteriálních zdrojů, jako například vektor YAC (yeast artificial chromosome) nebo HAC (human artificial chromosome).
Po úspěšném klonování a zavedení vektoru do vhodné hostitelské buňky je možné ke studiu získaných knihoven aplikovat dva přístupy – screening může být založený na sekvenování, nebo na funkční detekci klonovaných genů. Oba přístupy mají své výhody, ale také úskalí. Sekvenční screening je založen na detekci sekvencí podobných známým (funkčním) genům a může tedy detekovat (za použití primerů nebo sond) jen varianty již známých metabolických drah a podobně. Jejich detekce však nezajišťuje přítomnost celých takovýchto genů/drah. Výhodou tohoto přístupu je nezávislost na expresi a produkci cizích genů v hostitelské buňce. Případně je – díky nesmírnému pokroku v technikách sekvenování – teoreticky možné sekvenovat celé knihovny, kde pak největším problémem bude zpracování a především interpretace získaných výsledků. V případě funkčního screeningu není potřebná znalost jakékoliv sekvence a proto tímto přístupem mohou být objeveny zcela nové geny/funkce. Základním a limitujícím faktorem zde však bude exprese klonovaných genů a tvorba funkčních produktů v hostitelské buňce. Situace je dále komplikována faktem, že i v případě, že k této expresi a tvorbě produktů dojde, mohou tyto být pro buňku letální a v důsledku toho tedy nedetekovatelné. Tvorbě metagenomových knihoven a jejich analýze se budeme ještě podrobněji věnovat v následujících kapitolách práce.
Jako pro jiné aplikace v molekulární biologii i pro tvorbu metagenomových knihoven existují dnes i komerční kity. Například firma Epicentre vyvinula kit k izolaci DNA z vodného prostředí za účelem tvorby metagenomových knihoven (Metagenomic DNA Isolation Kit for Water, Epicentre, dnes součást Illumina, Inc., Madison, Wisconsin, USA). Uváděná velikost izolovaných fragmentů je však jen 40 kb, což může být podle našeho názoru nedostatečné. Firma Lucigen nabízí dokonce ucelený servis ve formě tvorby celých metagenomových knihoven: Metagenomic BAC and Fosmid Libraries. Podle informací na jejich webovských stránkách (http://lucigen.com/store/Metagenomic-BAC-and-Fosmid-Libraries.html) jsou schopni izolovat DNA o vysoké molekulové hmotnosti z minimálního množství nejrůznějšího materiálu (mikrobiální vzorky z rostlin a živočichů; půdní vzorky; mořské vzorky – plankton, řasy, houby; extremofilové; a jiné environmentální vzorky) a tuto pak použít k přípravě náhodně nastříhaných (Random Shear) BAC a fosmidových knihoven, které jsou zákazníkovi doručeny zmrazené v 384 nebo 96 jamkových destičkách. V době, kdy jsme se v našich laboratořích zabývali tvorbou metagenomových knihoven, žádný z těchto servisů neexistoval a proto jejich vhodnost nebo účinnost nemůžeme posoudit.
I přes všechny zmíněné problémy a omezení představují metagenomové knihovny významný a nepostradatelný prostředek k identifikaci nových biomolekul využitelných v medicíně (nová antibiotika a jiná léčiva), potravinářství i mnoha průmyslových aplikacích a v neposlední řadě i ke studiu diverzity a metabolického potenciálu mikrobiálních společenstev včetně dosud do značné míry opomíjených virových společenstev, pro které knihovny představují téměř jediný možný způsob jejich soustavného studia. Svědčí o tom i řada prací z poslední doby zabývajících se tvorbou a využitím metagenomových knihoven z půdního prostředí, jako například Nacke et al., 2011; Sang et al., 2011; Khan a Jithesh, 2012; Ko et al., 2012; McGarvey et al., 2012.
Metody studia mikrobiální diverzity založené na analýze R/DNA s použitím PCR
Polymerázová řetězová reakce
Až do počátku 80. let minulého století byla identifikace mikroorganismů i všechny navazující mikrobiologické metody založeny převážně na metodách kultivačních. Pak přišly dva významné objevy. V roce 1985 Kay Mullis pracující v laboratořích Henry Ericha (Cetus Corp. in Emeryville, Kalifornia) objevil polymerázovou řetězovou reakci. Druhým důležitým milníkem bylo zjištění, že fylogenetické vztahy mezi organismy mohou být odvozeny z molekulárních sekvencí. Zcela ideální se pro tento účel ukázal gen pro 16s rRNA, který splňuje všechny požadavky kladené na ideální molekulární marker: je přítomen ve všech živých organismech, je dostatečně dlouhý (první pokusy byly prováděny s kratší 5s rRNA), aby poskytl potřebné informace (dokumentace evoluce a podobně), jeho horizontální transfer je omezený, stejně tak jako jeho primární a sekundární struktura a konečně jeho sekvence obsahuje domény konzervativní (umožňující např. tvorbu primerů) i variabilní (umožňující rozlišení jednotlivých genů/organismů). Spojení těchto dvou objevů znamenalo doslova revoluci v mikrobiální ekologii i v celé mikrobiologii i ostatních biologických vědách. Vzhledem k významu PCR pro všechny biologické vědy se budeme touto metodou zabývat poněkud podrobněji.
Princip PCR spočívá v namnožení úseku nukleové kyseliny ohraničeného na začátku a na konci tzv. primery (krátkými oligonukleotidy DNA) v zařízení zvaném termocycler, které je schopné rychle měnit teplotu vzorků. Celý proces PCR pak sestává z mnohonásobného opakování kroků denaturace DNA (94oC), nasednutí primerů na oddělené jednovláknové řetězce denaturované templátové molekuly DNA (50–65oC) a prodlužování primerů – tedy syntéza komplementárního řetězce DNA (72oC). Teoreticky lze takto během 30 cyklů z jedné molekuly DNA získat přes miliardu identických molekul DNA.
Základním předpokladem pro úspěšnou aplikaci všech molekulárně biologických metod využívajících PCR je kvalitní pár primerů zabezpečující vysokou specificitu a účinnost PCR reakce. Proto všechny PCR metody začínají buď vyhledáváním vhodných již existujících primerů, nebo tvorbou nových primerů. Pro vytvoření nových primerů je nutné nejprve shromáždit maximum informací o DNA sekvenci zkoumané oblasti, tyto pak seřadit podle podobnosti sekvencí a na základě sekvencí konzervativních úseků DNA navrhnout vhodné primery.
V případě primerů pro 16S rDNA bude zdrojem potřebných informací nejpravděpodobněji databáze RDP – Ribosomal Database Project. Tato databáze vznikla na popud Carl R. Woese a Gary J. Olsena z University of Illinois, kteří si uvědomili, že rRNA, díky svým konzervativním sekvencím, může být využita ke zkoumání fylogenetických vztahů mezi organismy. Databáze obsahuje sekvence prokaryotické, eukaryotické a mitochondrialní malé podjednotky rRNA a nabízí i řadu služeb, jako seřazení dat dle sekvencí, tvorba dendrogramů, vyhledávání sekvencí dle podobností a podobně.
Kromě primerů pro 16s rRNA s různou specificitou (od univerzálních bakteriálních primerů až k druhově specifickým primerům) jsou také často využívány primery pro funkční geny. Princip jejich tvorby je podobný jako u 16s rRNA primerů, ale vzhledem k tomu, že se jedná o primery pro funkční geny, jejichž produktem jsou většinou enzymy či bílkoviny všeobecně, má jejich tvorba určitá specifika. Jde především o prvotní seřazení sekvencí zkoumané oblasti, které je možné udělat podobně jako u 16s rRNA primerů na úrovní bazí, ale v případě funkčních genů asi častěji na úrovni aminokyselin, které může odhalit některé skryté podobnosti mezi příbuznými sekvencemi.
Existuje celá řada programů pro tvorbu/designe primerů, na tomto místě zmíníme jen námi často úspěšně využívaný program CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primers), který je možné v původní formě nalézt na webové adrese http://blocks.fhcrc.org/blocks/help/CODEHOP/tips.html. Tento program už není v současné době dále udržován a je možné jej využít jen pro jednodušší úkoly/primery a byl nahrazen podobným programem iCODEHOP na Univerzity of Washington (http://dbmi-icode-01.dbmi.pitt.edu/i-codehop-context/Welcome). Do programu jsou nejprve vloženy původní aminokyselinové sekvence zkoumaného úseku DNA, případně přímo hotový soubor vzniklý seřazením sekvencí například pomocí programu ClustalW a tato data jsou předána na server. V případě větší podobnosti sekvencí program přímo navrhne soubor degenerovaných primerů (degenerovaný primer představuje směs primerů skládajících se z konzervované části společné pro všechny primery a části variabilní zabezpečující perfektní dosednutí primeru na maximální množství templátů v environmentálním vzorku), ze kterých je možné si vybrat nejvhodnější pár primerů. Pokud však jsou sekvence příliš různorodé a program není schopen navrhnout žádný primer, je nutné buď změnit podmínky tvorby primerů (v programu je například možné upřednostnit některé sekvence zvýšením jejich váhy/významu a podobně), případně zúžit výběr sekvencí (a tím i specificitu vytvořených primerů). Může dojít i k tomu, že i po změně podmínek pro dané sekvence není program schopen vytvořit žádné primery.
V případě, že jsou primery nalezeny, je vhodné (nutné) jejich specificitu ověřit nejprve in silico, tedy porovnáním jejich sekvence se známými sekvencemi v databázích, což se většinou děje za pomoci programu BLAST (The Basic Local Alignment Search Tool), http://blast.ncbi.nlm.nih.gov/Blast.cgi. Pokud se prokáže dostatečná specificita, je možné přistoupit k optimalizaci podmínek PCR reakce a ke kontrole specificity primerů s využitím co možná největšího souboru DNA z cílových a příbuzných mikroorganismů (kde je očekáván PCR produkt), ale i „cizích“ organismů (kde naopak by nemělo k tvorbě PCR produktů dojít). Příklad optimalizace PCR reakce prováděný v naší laboratoři pro nové primery (podobná optimalizace by měla být prováděna vždy při jakékoliv změně v PCR reakci – například i při změně dodavatele DNA polymerázy) uvádí Tab. 3. Údaje v tabulce vychází z následujících předpokladů: všechny reakce jsou prováděny v konečném objemu 25 µl; konečná koncentrace dNTPs je 200 µM; koncentrace MgCl2 vychází z předpokladu, že je k taq polymeráze dodáván pufr bez MgCl2; v případě, že je MgCl2 součástí dodávaného PCR pufru, bude první koncentrace MgCl2 nulová a další dvě mohou být vhodně upravené; zásobní roztok BSA – bovinní sérový albumin – je připraven v koncentraci 10 mg/ml a do reakce je přidáván za účelem odstranění inhibicí z environmentální templátové DNA (především huminové látky); DMSO – dimethyl sulfoxide – do reakce je přidáván pro zvýšení její účinnosti zejména usnadněním kompletní denaturace templátové DNA; primery jsou naředěny tak, aby 1 µl obsahoval 50 pmolů primeru; vzhledem k tomu, že u většiny PCR reakcí v naší laboratoři využíváme horký start (hot start), je naředěná taq polymeráza přidávána po počáteční denaturaci do každé zkumavky zvlášť (na 50 µl reakci připadá 1 U polymerázy); SDW – sterilní destilovaná voda.
| Komponenty | ||||||||||||
| dNTPs | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| PCR buffer | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| MgCl2 | 3 | 6 | 9 | 3 | 6 | 9 | 3 | 6 | 9 | 3 | 6 | 9 |
| BSA | 10 | 10 | 10 | 10 | 10 | 10 | – | – | – | – | – | – |
| DMSO | 5 | 5 | 5 | – | – | – | 5 | 5 | 5 | – | – | – |
| Primer 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Primer 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Taq polymeráza | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 | 8 |
| DNA | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| SDW | 59 | 54 | 49 | 64 | 59 | 54 | 69 | 64 | 59 | 74 | 69 | 64 |
| Celkem | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| dNTPs – 10mM; MgCl2 – 50 mM; BSA – 10mg/ml; primery – 50 pmolů v 1 μl; taq polymeráza – 0,5 U/ μl | ||||||||||||
Po připravení všech 12 směsí (mastermixů) jsou tyto rozpipetovány do 4 zkumavek, z nichž tři slouží pro přidání templátové DNA z organizmů, pro které byl primer připraven, čtvrtá je použita jako negativní kontrola. Je také možné použít méně vzorků templátové DNA a místo toho zbývající alikvoty mastermix využít pro testování PCR podmínek při jiné teplotě přiložení primerů (annealing temperature). Jak vyplývá z tabulky, při optimalizaci jde především o určení vhodné minimální koncentrace MgCl2, která rozhoduje o specificitě reakce. U ostatních komponent – především BSA a DMSO – jde o zjištění vlivu jejich (ne)přítomnosti na průběh reakce. Zvyšováním koncentrace MgCl2 přibývá PCR produktu, ale vzhledem ke snižování specificity reakce (umožněním dosednutí primerů na úseky DNA s menší shodou s primerem) se zvyšuje nebezpečí vzniku nespecifických produktů. Podobný efekt má snižování teploty přisedání primerů (annealing temperature), který lze – místo změn koncentrace MgCl2 – s výhodou testovat na gradientových PCR cyclerech. Výsledkem optimalizace PCR reakce by měl být PCR produkt o požadované velikosti, jehož specificitu si lze ověřit buď sekvenováním, nebo například hybridizací s vhodnou sondou.
I přes nezpochybnitelný význam PCR má tato metoda některé problémy, se kterými je nutno počítat a které zde v krátkosti zmíníme. V prvé řadě funkce DNA polymeráz používaných pro syntézu nových řetězců DNA není bezchybná. Běžně používaná taq polymeráza (izolovaná z bakterie Termus aquaticus) je sice účinná, postrádá ale tzv. „3' to 5' exonuclease proofreading aktivity“, tj. schopnost kontrolovat správnost zkopírované DNA. To znamená, že 1 z 9000 nukleotidů zabudovaných do nově syntetizované molekuly DNA je chybný. Vznikne tedy mutovaná molekula DNA, která je během dalších cyklů PCR dále amplifikovaná. Pfu polymeráza (izolovaná z Pyrococcus furiosus) kontrolní (proofreading) aktivitu má a četnost zabudování chybného nukleotidu u této polymerázy je pouze 1 v 1,3 miliónech bazí, je však pomalejší a proto se využívá především tam, kde jsou vysoké nároky na přesnost replikace DNA (klonování a podobně).
Další příčinou vzniku mutovaných molekul je přítomnost sekundárních struktur na amplifikované DNA. V důsledku takovýchto struktur může dojít při kopírování DNA k přeskočení krátkého úseku DNA, tedy k deleci, jejímž výsledkem je kratší PCR produkt, tedy opět „mutace“ dál množená v následujících cyklech PCR.
Závažným, často přehlíženým problémem, je vznik tzv. chimér, tedy nových molekul DNA, které se nenacházely v původním vzorku/templátu. K tomuto jevu dochází tehdy, když z jakéhokoliv důvodu nedojde v jednom cyklu k dokončení syntézy kopírovaného úseku DNA. Při dalším cyklu může tato krátká molekula DNA nasednout na jinou molekulu DNA, se kterou je v určitém úseku (i odlišném od okrajových oblastí, kde dosedají primery) homologní a je potom dosyntetizována podle tohoto templátu. Vzniklá molekula DNA se pak skládá ze sekvencí ze dvou různých templátů a je tedy zcela nová, v původním vzorku se nevyskytující. Některé zdroje uvádí, že za určitých okolností (například přítomnost malých fragmentů DNA v templátové DNA, které mají za následek předčasné ukončení syntézy DNA v jednom cyklu a její dosyntetizování podle příbuzného ale odlišného templátu v cyklu následujícím; dále přítomnost sekvencí s vysokým procentem konzervovaných úseků DNA, kdy místo primerů k sobě dosednou jiné molekuly DNA a jsou pak chybně dosyntetizovány) mohou chiméry tvořit až 30 % molekul PCR produktu. Pokud by tomu tak bylo, význam PCR reakce pro molekulární biologii by byl významně zpochybněn. Vzniku chimér se lze bránit používáním kvalitní templátové DNA (DNA o vysoké molekulové hmotnosti), prodloužením doby prodlužování primerů (extension) a snížením počtu cyklů PCR. Kromě toho byly vytvořeny programy na detekci chimér, které však spolehlivě fungují jen za předpokladu, že obě sekvence (templátové molekuly DNA) jsou odlišné minimálně 15 %. K odhalení chimér je nutné provádět důkladnou analýzu produktů, jejich sekundární struktury, fylogenetické analýzy různých částí molekul, či sestavit dendrogramy z opačných konců molekul, které – v případě chimér – budou odlišné.
Dalším problémem PCR je tzv. diferenciální amplifikace. Teoreticky by měly být v každém cyklu PCR replikovány všechny přítomné templátové molekuly DNA, což by znamenalo, že ve výsledném produktu budou zachovány přesně původní poměry ve vzorku. Pracujeme-li však například s mikrobiální DNA izolovanou z environmentálního vzorku a používáme primery pro 16S rRNA, může dojít k tomu, že vzhledem k odlišnému počtu kopií rDNA v geonomu různých bakterií, budou ve výsledném produktu převažovat sekvence z bakterií s vyšším počtem těchto genů a takto dojde ke zkreslení původního složení vzorku. Je také třeba vzít v úvahu heterogenitu rRNA operonů v jednotlivých bakteriích, které mohou mít za následek vytvoření umělé diverzity vzorku (dva různé operony genu 16S rRNA ze stejného organismu budou pokládány za molekuly pocházející ze dvou různých organismů).
Ke zkreslení zastoupení jednotlivých sekvenci představujících různé mikrobiální populace zastoupené v původním vzorku/komunitě může dojít také v případě, že jsou v něm velké rozdíly v početním zastoupení jednotlivých populací mikroorganismů. S přibývajícím počtem cyklů klesá koncentrace primerů a stoupá koncentrace produktů, takže v pozdějších cyklech nemusí vždy dojít k nasednutí primeru na templátovou DNA, ale místo toho může docházet k opětnému spojení dvou templátových molekul (tzv. re-annealing). V tomto případě templát v nadbytku (majoritní/dominantní populace) dosáhne dřív kritické koncentrace, kde pak převládne opětné dosedání templátových molekul k sobě (re-annealing) namísto dosedání primerů na templátovou molekulu a tato DNA se pak již nebude amplifikovat a její množství bude stagnovat, zatímco minoritní templátová DNA (minoritní populace) bude dále amplifikována, takže ve výsledném PCR produktu budou setřeny původní rozdíly v zastoupení jednotlivých populací mikroorganismů.
Z dalších možných příčin diferenciální amplifikace je třeba zmínit rozdíly v obsahu G+C templátu (templáty s vyšším obsahem G+C vzhledem k pevnější vazbě mezi C-G oproti A-T mohou hůře denaturovat a v důsledku toho mohou být amplifikovány s nižší účinností ve srovnání s templáty s vyšším obsahem A+T), přítomnost sekvencí (i mimo amplifikovaný úsek), které snižují účinnost amplifikace (smyčky/loops interferující s prodlužováním/elongací – extension), nebo modifikace templátu (metylace) zabraňující amplifikaci.
Velký význam má také účinnost/efektivita a selektivita primerů – suboptimální vazba primerů sníží účinnost amplifikace; univerzální primery mohou zapříčinit preferenční amplifikaci některých sekvencí nejpodobnějších primerům; degenerované primery (směs primerů s různou sekvencí) tuto chybu mohou odstranit, ale pokud se liší teplota tání (Tm) jednotlivých primerů, různá teplota nasedání primerů bude opět příčinou diferenciální amplifikace.
Nicméně i přes všechny výše zmíněné problémy přínos této metody přinejmenším vyvažuje zmíněné nedostatky a PCR je proto využívána v řadě molekulárně biologických metod určování diverzity mikrobiálních společenstev, jejichž stručný přehled následuje.
Polymorfismus délky restrikčních fragmentů (RFLP, T-RFLP, ARDRA)
RFLP, restriction fragment length polymorphism, polymorfismus délky restrikčních fragmentů (nebo také ARDRA, Amplified ribosomal DNA restriction analysis), byla první technika analýzy DNA, která doznala významného rozšíření. Při této technice je analyzovaná DNA štěpena restrikčními endonukleázami a vzniklá směs DNA fragmentů rozdělena pomocí elektroforézy, v základní verzi v agarózovém gelu. Poté může za účelem identifikace fragmentů následovat přenos DNA na membránu a hybridizace se značenou sondou. Pomocí RFLP lze od sebe odlišit dvě (a více) příbuzné/homologní molekuly DNA lišící se v jednom či více párech bazí (bodová mutace, inzerce, delece, inverze, translokace) za předpokladu, že tato odlišnost se nachází buď v místě rozeznávaném použitým restrikčním enzymem, nebo jí dojde ke změně délky restrikčního fragmentu DNA detekovatelné elektroforeticky (pro vyšší citlivost je možné/nutné nahradit elektroforézu v agarózovém gelu elektroforézou v polyakrylamidovém gelu). Tato metoda sehrála významnou roli v mapování geonomů a genetické analýze nemocí, byla používána k vytváření prvních genetických fingerprintů, určování paternity, v kriminalistice a k určování genetické diverzity populací (Walker-Jonah et al., 1992; Echt et al., 1994; Umene a Yoshida, 1994; Wu et al., 2009; Chuah et al., 1994).
K charakterizaci diverzity mikrobiálních společenstev vyvinuli Liu et al (1997) modifikaci této metody, tzv. T-RFLP, polymorfismus délky terminálních restrikčních fragmentů, který je využíván jak pro ribozomální geny, tak i pro jiné funkční geny (Boyle-Yarwood et al., 2008; Wu et al., 2009; Yarwood et al., 2010). V tomto případě je environmentální DNA použita jako templát pro PCR reakci, kde jeden nebo oba primery jsou fluorescenčně značené a po proběhnuté PCR a štěpení vhodnými endonukleázami a elektroforéze jsou na gelu viditelné pouze terminální restrikční fragmenty (Obr. 3-2). Výsledkem je zjednodušený/zpřehledněný fingerprint mikrobiálního společenstva, kde je každý druh reprezentován jen jedním nebo dvěma (při značení obou primerů) fragmenty. Detekce fragmentů se v tomto případě většinou provádí na DNA sekvenátoru, který je schopen rozlišit fragmenty lišící se délkou jednoho nukleotidu. Výsledný elektroforegram podá kvalitativní (počet jednotlivých druhů reprezentovaných jednotlivými píky) i semikvantitativní (výška/intenzita píku odráží do určité míry množství jedinců téhož druhu) informaci o složení společenstva (Obr. 3-3).

Obr. 3-2 Princip techniky T-RFLP
(převzato z http://nodens.ceab.csic.es/t-rfpred/method; Fernàndez-Guerra et al., 2010)
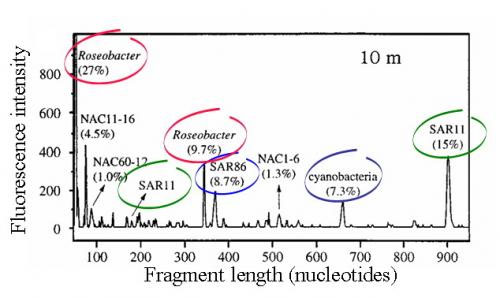
Obr. 3-3 Příklad analýzy mikrobiálního společenstva pomocí T-RFLP
(převzato z http://nodens.ceab.csic.es/t-rfpred/method; Fernàndez-Guerra et al., 2010)
V porovnání s ostatními fingerprintovými metodami, především s T/DGGE, má tato metoda některé klady i zápory. K výhodám patří použití automatického sekvenátoru, které značně zvyšuje reprodukovatelnost výsledků, které jsou navíc přímo produkovány v digitální formě vhodné k dalšímu, především numerickému zpracování i jednoduchému skladování. K nevýhodám patří především fakt, že každý druh je zastoupen pouze terminálním restrikčním fragmentem, kde je zvýšená pravděpodobnost, že příbuzné druhy nebudou touto technikou rozlišeny (budou mít společný terminální fragment). Další nevýhodou je nemožnost získat sekvenci a tedy přesnější identifikaci detekovaných píků.
Gelová elektroforéza v teplotním/denaturačním gradientu (T/DGGE)
Jednou za základních tzv. fingerprintových metod je DGGE nebo TGGE, tedy Denaturing gradient gel electrophoresis, nebo Temperature gradient gel electrophoresis, česky gelová elektroforéza v denaturačním gradientu nebo gelová elektroforéza v teplotním gradientu. Princip této metody byl známý již dlouho před její aplikací pro environmentální mikrobiologii, kdy byl využíván pro detekci bodových mutací DNA (metoda je v principu schopná odlišit od sebe dvě sekvence lišící se jedním párem nukleotidů). Nová byla její aplikace na gen 16S rDNA spolu s přidáním tzv. GC svorek, tak jak tuto metodu v roce 1993 publikovali Muyzer et al. Princip obou metod (DGGE i TGGE) je shodný. PCR produkt migruje v polyakrylamidovém gelu s denaturačním gradientem, který je buď chemický (močovina a formamid), nebo teplotní. Ve skutečnosti jde v obou případech o kombinaci obou přístupů – tedy denaturace je dosaženo kombinací zvýšené teploty a denaturačních chemikálií a rozdíl spočívá jen v tom, čím je tvořen vlastní gradient – buď tedy u DGGE gradient chemický – gradient koncentrace močoviny a formamidu, kdy elektroforéza probíhá za zvýšené teploty (65oC), nebo gradient teplotní u TGGE, kdy je v gelu jednotná koncentrace formamidu a močoviny. Zpočátku molekuly DNA migrují gelem rychle, ale se zvyšujícím se denaturačním gradientem dochází k postupné denaturaci molekul DNA, oddělování řetězců DNA, což snižuje rychlost jejich postupu gelem. Průběh této denaturace je specifický dle sekvence analyzované DNA. Vazby mezi G a C se denaturují pomaleji než vazby mezi A a T. V konečném důsledku by došlo k úplnému oddělení dvoušroubovice DNA a vyplavení rychle migrující jednořetězcové DNA z gelu, čemuž je zabráněno přítomností tzv. GC svorky, tedy úseku dlouhého zhruba 40 párů bazí skládajícího se téměř výhradně z G a C, který je k analyzovanému úseku přidán (je součástí jednoho z primerů), drží oba řetězce DNA u sebe a má za následek úplné zastavení postupu analyzované molekuly v gelu (Obr. 3-4). To znamená, že při analýze mikrobiální komunity (pomocí 16/18S rDNA, nebo některého funkčního genu) je výsledkem řada proužků, tzv. fingerprint, protože teoreticky každý mikrobiální druh má svou specifickou sekvenci rDNA (případně odlišnou sekvenci funkčního genu) a tomu odpovídá specifická poloha proužku na gelu (Obr. 3-5). Tedy opět teoreticky každý mikrobiální druh dá vzniknout jednomu proužku na gelu a naopak, každý proužek reprezentuje jeden mikrobiální druh. Slovo teoreticky jsme zdůraznili proto, že jde opravdu jen o teoretický předpoklad, který ve většině případů také platí, nicméně bylo již prokázáno, že dva organismy mohou dát vzniknout proužku se stejnou polohou na gelu a proto také jeden proužek může odpovídat dvěma a více mikroorganismům (Muyzer a Smalla, 1998) a konečně jeden organismus může (z důvodu přítomnosti více kopií zkoumaného genu – např.16S rDNA) dát vzniknout více proužkům (Nubel et al., 1996).
Úspěšnost metody je závislá v prvé řadě na vhodném výběru analyzované DNA sekvence, která musí být dostatečně variabilní, aby opravdu každý druh nebo jiná nomenklaturní jednotka byla reprezentována jedinečnou sekvencí, která umožní její rozlišení na gelu. Nicméně ani potom není zaručeno, že je bude možné na T/DGGE odlišit, tak jak již bylo řečeno. Lze si dobře představit situaci, kdy dva co do sekvence odlišné úseky DNA dají při denaturaci vzniknout prostorově shodné sekundární či terciární struktuře se shodnou migrací v polyakrylamidovém gelu a vytvoří tedy
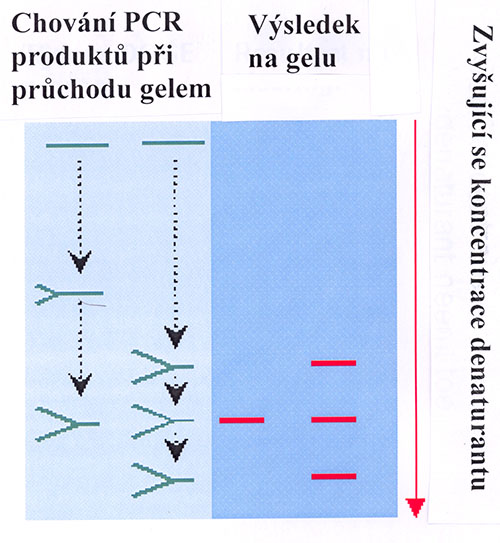
Obr. 3-4 Princip techniky DGGE
Obr. 3-5 DGGE analýza mikrobiálního společenstva zažívacího traktu včel; jednotlivé běhy představují mikrobiální komunity zažívacího traktu včely medonosné u vzorků odebraných ze včel různého stáří z několika lokalit.
splývající proužek na tomto gelu. Je také třeba mít na paměti, že fingerprint komunity bude reprezentovat jen nejčetnější představitele komunity, tj. mikrobiální druhy s nízkým počtem jedinců v dané komunitě nebudou s největší pravděpodobností na gelu reprezentovány proužkem (vytvoří na gelu tzv. „smear“, tedy česky šmouhu, či šum nebo pozadí gelu). MacNaughton et al. (1999) odhadovali, že pomocí DGGE lze detekovat 1–2 % mikrobiální populace představující dominantní druhy v daném vzorku. Toto číslo se nápadně blíží odhadu podílu společenstva, který lze analyzovat pomocí tradičních kultivačních metod. Je tu ale zásadní rozdíl v tom, že u kultivačních metod se jedná o cca 1 % mikroorganismů schopných růstu na kultivačním médiu bez ohledu na jejich úlohu a procentuální zastoupení v dané komunitě, zatímco u T/DGGE jde o 1–2 % nejpočetnějších mikroorganismů, o kterých se dá předpokládat, že vzhledem ke svému početnímu zastoupení také plní klíčové role ve společenstvu. Je samozřejmé, že se snižující se diverzitou společenstva podíl jejich členů detekovaných T/DGGE stoupá. Kromě toho existují postupy, jak lze s pomocí T/DGGE detekovat i minoritní populace. V principu lze rozlišit dva přístupy; buď lze aplikovat určitou selekci ještě před vlastním PCR, nebo samo PCR může být selektivní (primery, nested PCR).
Selekce před PCR může být provedena ještě před vlastním odběrem vzorku, nebo až po izolaci DNA. Použití techniky tzv. „burried litter bags“, tedy zakopaných sáčků se substrátem, je příkladem prvního přístupu (Krsek a Wellington, 2001; Metcalfe et al., 2002a; Aneja et al., 2004). V tomto případě je vybraný substrát (většinou uzavřený v sáčku z nylonové nebo podobné síťoviny připomínající čajové sáčky) aplikován do přirozeného prostředí a po určité době expozice/kultivace vyjmut a mikroflóra kolonizující tento substrát podrobena analýze (Obr. 3-6). Tímto způsobem je možné detekovat i minoritní populace, které by při analýze vzorku získaného přímo z přirozeného prostředí vzhledem k nízkému zastoupení v mikrobiálním společenstvu mohly být pod limitem detekce zvolené metody. Je ovšem třeba mít na paměti, že jde o druh obohacovací kultury (podobného efektu je možné dosáhnout jen prostým obohacením prostředí vybraným substrátem), která výrazně změní původní zastoupení jednotlivých mikrobiálních populací ve vzorku.
Druhou možností je selekce na úrovni izolované DNA. I zde se nabízí více možností, všimněme si alespoň dvou z nich. V obou případech jde o frakcionaci DNA. Jednou z možností je rozdělení DNA na základě obsahu G+C za pomocí barviva bis-benzimidazole. Toto se přednostně váže k sekvenci AT a takto zvyšuje hustotu (buyoan density) DNA, kterou lze pak rozdělit v gradientu chloridu cesného. Využitím takto rozdělené DNA lze dosáhnout větší citlivosti při vlastní DGGE. Tento princip využili například Holben a Harris (1995) k monitorování změn ve složení půdní mikrobiální komunity v závislosti na změnách obsahu uhlíku a vody v prostředí a o téměř desetiletí později (Holben et al.,2004) v kombinaci s DGGE ke studiu minoritních mikrobiálních populací mikroflóry zažívacího traktu kuřat. Specifická vazba barviva bis-benzimidazole byla využita i pro jednoduché rozlišní rozdílných sekvencí dvou druhů Desulfovibrio při elektroforéze v agarózovém gelu s přídavkem tohoto barviva (Wawer et al., 1995).

Obr. 3-6 Ukázka techniky zakopaných sáčků se substrátem (burried litter bags)
Jinou možností je již výše zmíněné značení cílových molekul DNA těžkými stabilními izotopy prvků, nejčastěji 13C (Radajewski et al., 2000). V tomto případě probíhá selekce na základě využití značeného substrátu cílovou skupinou mikroorganismů. Jejich DNA bude po zabudování těžkých izotopů prvků těžší a bude ji možno oddělit od ostatní DNA opět v gradientu chloridu cesného. DGGE fingerprint připravený za použití takto získané DNA bude potom reprezentovat pouze mikroorganismy využívající daný substrát. Jak již bylo zmíněno výše, má tato metoda některá úskalí. V prvé řadě není vždy jednoduché získat určitý substrát značený těžkým prvkem (jeho výběr je kromě toho omezen prvky tvořícími DNA) a pokud je takový substrát k dispozici, je téměř vždy velice drahý. Dalším problémem metody je možnost tzv. crossfeedingu, tedy posun značeného prvku potravním řetězcem (čehož – na druhé straně – může být někdy naopak využito právě pro studia zmíněného potravního řetězce). K dosažení dostatečného značení DNA je totiž nutné podat značné množství značeného substrátu a dodržet určitou dobu inkubace, aby byl značený prvek zabudován do DNA. Během této doby už ale také může dojít k rozkladu časti značené populace, či exkreci části značeného prvku ve formě nejrůznějších metabolitů a k přijetí těchto substrátů jinými mikroorganismy přítomnými ve sledované komunitě. Kromě toho může nastat i problém s příjmem zvýšeného množství těžkých izotopů prvků organismy. Aplikace této techniky (tzv. SIP – stable izotope probing) díky její náročnosti (drahé substráty) i technickým problémům byla původně omezená především na specializované komunity, kde například možnost crossfeeding je značně omezená (metanotrofní mikroorganismy – Radajewski et al., 2000), nicméně postupně se začaly využívat i při studiu jiných komunit. Například Taubert et al. (2012) použili tuto techniku ke studiu síru redukujících a benzen degradujících mikrobiálních společenstev, nebo Xia et al. (2011) publikovali článek, ve kterém použili SIP techniku ke studiu amonium oxidujících bakterií a archeí v zemědělské půdě.
Druhý způsob detekce minoritních populací může být založen na specificitě PCR samotné. Úspěšnost/proveditelnost tohoto přístupu závisí na tom, zda existují, nebo je možné vytvořit, selektivní primery specifické pro zkoumanou skupinu mikrobů. V kladném případě je pak možné přímo aplikovat tyto primery a získat fingerprint zkoumané skupiny mikrobů, případně je možné použít tzv. hnízdovou PCR (nested PCR), kdy v prvním kole PCR je využita dvojice univerzálních primerů a teprve ve druhé PCR, kde jako templátová DNA slouží PCR produkt první reakce, jsou aplikovány specifické primery (které však musí ležet uvnitř prvního PCR produktu). V prvním PCR dojde k namnožení cílových sekvencí DNA (včetně sekvencí z minoritních populací) a v druhém kole pak k samotné detekci minoritních populací (Heuer et al., 1997; Dar et al., 2005; Mahmood et al., 2006).
I přes všechny výše zmíněné nedostatky či limity došly metody T/DGGE nejširšího uplatnění v environmentální mikrobiologii. Za použití 16s rDNA genů, ale i funkčních genů, byly použity ke stanovení diverzity nejrůznějších mikrobiálních společenstev, ke sledování změn v jejich složení v čase i v prostoru (Dar et al., 2005; Bodelier et al., 2005; Mahmood et al., 2006; Campbell et al., 2009). Jde o metodu spolehlivou, reprodukovatelnou, rychlou a relativně levnou.
Polymorfismus konformace jednořetězcových nukleových kyselin (SSCP)
Podobný princip jako T/DGGE, tedy princip konformační změny molekuly DNA v závislosti na její sekvenci, využívá i metoda SSCP, tedy „single-strand conformation polymorphism“, polymorfismus jednořetězcových nukleových kyselin. Tato metoda využívá faktu, že elektroforetická mobilita jednořetězcové DNA není na rozdíl od dvouřetězcové DNA uniformní (závislá pouze na její velikosti), nýbrž závislá na její sekvenci. Za nepřítomnosti komplementárního řetězce bude docházet k párování bazí v rámci téhož řetězce a výsledkem bude specifická trojrozměrná struktura tohoto řetězce, která ovlivní jeho elektroforetickou mobilitu. Výsledkem elektroforézy bude tedy opět jistý fingerprint analyzované komunity (podobný fingerprintům/gelům DGGE). V porovnání s předešlými metodami, především T/DGGE, se jedná o jednodušší metodu nevyžadující GC svorky ani gradientové gely. I tato metoda má však své problémy. Jedním z nich je fakt, že jeden PCR produkt může dát vzniknout hned třem různým trojrozměrným strukturám – dvě pro každý řetězec DNA a třetí pro dvouřetězcovou DNA. Tomuto se dá zabránit odstraněním jednoho z řetězců (například výrazným přebytkem jednoho z primerů při PCR, nebo odstraněním jednoho z řetězců – fosforylovaného – exonukleázovou aktivitou). Dalším problémem je závislost mobility jednořetězcové DNA na teplotě – elektroforéza musí probíhat za konstantní teploty. A konečně citlivost metody je závislá na pH a na velikosti analyzovaného fragmentu. Z těchto důvodů nedoznala tato metoda rozšíření srovnatelného s ostatními fingerprintovými metodami (DGGE, T-RFLP). Byla využita například pro analýzu změn v bakteriálních komunitách při kompostování (Peters et al., 2000), nebo k rozlišení mikroorganismů z čistých kultur (Medlin et al., 2006), k porovnání rozdílů dvou rhizosferních mikrobiálních komunit (Schwieger a Tebe, 1998), i ke studiu půdních houbových společenstev (He et al., 2005).
Analýza polymorfismu délky intergenového spaceru mezi geny malé a velké ribozomální podjednotky – Ribosomal Intergenic Spacer analysis – RISA
Metoda RISA je založena na PCR amplifikaci oblasti rRNA operonu rrl mezi geny pro malou (16S) a velkou (23S) podjednotkou zvanou intergenic spacer region (ISR). Použité primery využívají konservativních oblastí genů 16S a 23S. Význam ISR oblasti je založen na faktu, že tato oblast vykazuje významnou heterogenitu délkovou i sekvenční využitelnou pro taxonomické účely. RISA využívá délkovou variabilitu oblasti uváděnou v rozsahu 50 – 1500 bp (Ranjard et al., 2001). Výsledkem PCR reakce za použití environmentální DNA je směs fragmentů o různé délce odvozených z jednotlivých členů společenstva, která je následně rozdělena elektroforeticky na polyakrylamidovém gelu. Původní metoda byla vylepšena fluorescenčním značením jednoho z primerů (podobně jako metoda T-RFLP), kde pak k vizualizaci výsledků může být využit sekvenátor a metoda je touto formou automatizována – tzv. ARISA (automated RISA). Podobně jako u jiných metod může být v tomto případě odvozena z výšky píků i semikvantitativní informace o zastoupení jednotlivých skupin organismů ve společenstvu. V porovnání s ostatními výše uváděnými metodami (RFLP, T/DGGE, SSCP) se jedná o velice rychlou a jednoduchou metou nevyžadující žádné speciální vybavení (samozřejmě v její původní neautomatizované formě – RISA), denaturační gely ani štěpení restrikčními enzymy produkující fingerprint zkoumaného společenstva. Podobně jako například u T/DGGE i dalších metod mohou být proužky gelu separovány a sekvenovány, zde však ve srovnání s například již zmíněnou metodou T/DGGE bude u metody RISA hodnocení získaných výsledků obtížnější, protože velikost ISR databáze není srovnatelná s RDP databází. Také vysoká variabilita v délce zkoumané ISR oblasti zvyšuje nebezpečí diferenciální amplifikace, kdy jsou přednostně amplifikovány kratší sekvence. Nicméně již zmíněná jednoduchost a rychlost předurčuje tuto metodu k využití přinejmenším v laboratořích postrádajících speciální výbavu potřebnou pro jiné molekulární fingerprintové metody. V nedávné minulosti byla metoda využita například pro sledování změn struktury mikrobiálních komunit v PCB kontaminovaných půdách (Petric et al., 2011a), spolu s klonováním a sekvenováním 16S rRNA k monitorování mikrobiálních komunit v půdě při uplatnění různých sledů rostlin při pěstování arašídů (Sudini et al., 2011), nebo k hodnocení variability výsledků výše zmíněné metody izolace půdní mikrobiální DNA – ISO standard 11063 "Soil quality – Method to directly extract DNA from soil samples" (Petric et al., 2011b).
Délková heterogenita PCR produktů (LH PCR – Length Heterogeneity PCR)
Při tradičním pojetí PCR se předpokládá, že za použití jednoho páru PRC primerů vznikne produkt o stejné délce dané vzdáleností obou primerů na templátové DNA, který je detekovatelný po elektroforéze v agarózovém gelu jako jeden proužek. Tento předpoklad je platný použijeme-li jako templát DNA izolovanou z čisté bakteriální kultury (i zde může teoreticky vzniknout více odlišných PCR produktů, pokud je v genomu zkoumaného organismu přítomno více odlišných kopií cílového genu). Použijeme-li jako templát DNA izolovanou z přirozeného mikrobiálního společenstva, bude výsledný PCR produkt s největší pravděpodobností na agarózovém gelu také představovat jeden proužek. Podrobíme-li však tento PCR produkt detailnější délkové analýze (např. na sekvenátoru), zjistíme, že se ve skutečnosti jedná o směs produktů přibližně stejné délky (ve speciálních případech – viz např. výše uvedená délková heterogenita operonu rrl mezi geny pro malou (16S) a velkou (23S) podjednotkou využívaná metodou RISA – mohou být rozdíly délky velice významné). Toto je způsobeno skutečností, že geny kódující stejnou funkci/vlastnost nejsou v různých organismech zcela identické a s rostoucí fylogenetickou vzdáleností organismů roste i odlišnost těchto genů. V případě LH-PCR se odlišností rozumí délka genu, který například díky delecím či inzercím je buď kratší, nebo delší. Použijeme-li fluorescenčně značené PCR primery a podrobíme-li výsledný PCR produkt analýze na sekvenátoru, výsledkem pak bude elektroforegram, kde jednotlivé píky představují skupiny ve většině případů blízce „příbuzných“ mikroorganismů s velice podobnou/stejnou délkou PCR produktu zkoumané oblasti DNA (Obr. 3-7). Například Ritchie et al. (2000) tuto metodu použili ke srovnání 4 odlišných půd lišících se půdním typem nebo historií pěstování rostlin a prokázali, že tato metoda, podobně jako FAME, byla schopna odlišit srovnávané půdy.
Tuto metodu jsme testovali (Obr. 3-7) v rámci práce na EU projektu ACTAPHARM, avšak vzhledem k tomu, že jsme v té době ke studiu diverzity mikrobiálních společenstev již rutinně používali metodu DGGE, neshledali jsme ji přínosnou. Podle našeho názoru může najít uplatnění pouze v laboratořích vybavených klasickým Sangerovým sekvenátorem avšak postrádajících potřebnou výbavu pro jiné fingerprintové metody, jako zmíněné T/DGGE, RFLP a podobně. I z ekonomického hlediska je těžké si představit, že by se dal ospravedlnit nákup sekvenátoru pouze za účelem aplikace LH PCR, když mnohem levněji se dá zakoupit T/DGGE, s jehož pomocí se dají získat mnohem komplexnější a podrobnější výsledky při analyzování mikrobiálních komunit.
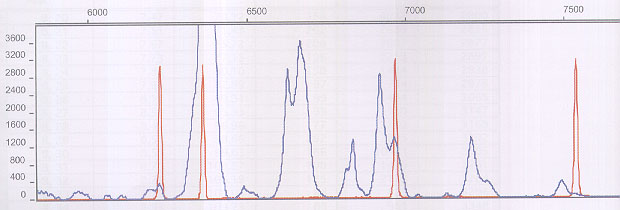
Obr. 3-7 Příklad LH-PCR profilu půdy za použití PCR primerů specifických pro geny syntézy polyketidů (EU projekt Actapharm); červené píky jsou délkové standardy a modré představují zkoumanou mikrobiální komunitu
Náhodná amplifikace polymorfní DNA (RAPD, AP-PCR)
Metoda RAPD (Randomly amplified polymorphic DNA) využívá jednoho náhodného krátkého PCR primeru (kolem 10 bp), který za nepříliš přísných podmínek PCR dosedá na jemu podobná místa na DNA vzorku. V případě, že tato místa jsou nedaleko sebe na protilehlých řetězcích DNA, dojde k vytvoření krátkého PCR produktu a každému přítomnému genomu pak odpovídá několik (5–15) takovýchto amplikonů (Franklin et al., 1999). V porovnání s ostatními diskutovanými metodami nedává RAPD příležitost k získání mnoha taxonomických informací o složení studovaného společenstva a lze ho využít především k prostému konstatování změn v tomto složení (téměř bez jakékoliv specifikace). Vzhledem k možnému velkému počtu produkovaných amplikonů by ho také nebylo vhodné využít ke studie komplexních mikrobiálních společenstev. Na druhé straně se jedná o jednoduchou, rychlou a především levnou metodu bez zvláštních nároků na technické vybavení. Metoda byla využita pro sledování složení akvatických komunit (Franklin et al., 1999), k charakterizaci izolátů aktinobakterií z antarktických půd (Lee et al., 2012), nebo k hodnocení metagenomového složení půd při hledání vhodných mikroorganismů pro účely bioremediace půd po petrochemické kontaminaci (Amorim et al., 2012).
Kvantitativní PCR
Nedílnou součástí analýzy diverzity mikrobiálního společenstva je i určení kvantitativního zastoupení jednotlivých druhů, případně taxonomických skupin. Přímé využití PCR produktů za tímto účelem (například kvantifikace na základě intenzity DGGE bandů) není vhodné díky již zmíněné diferenciální amplifikaci a je ho možné považovat v nejlepším případě za semikvantitativní. Existují však možnosti, jak této kvantifikace dosáhnout. Zhang a Xu (2008) uvádí čtyři možnosti kvantitativní PCR: „limiting dilution PCR“, kinetické PCR, kompetitivní PCR a real-time PCR. V případě „limiting dilution PCR“ je provedeno více PCR reakcí s postupně ředěnou templátovou DNA a pomocí tabulek je pak možné odhadnout původní koncentraci templátu. Kinetická PCR vyžaduje standardní templát známé koncentrace a kvantifikace je založena na zvýšení počtu amplikonů templátové a standardní DNA na jeden PCR cyklus. Je ale třeba podotknout, že název kinetická PCR se často také používá pro označení Real-Time PCR. Při kompetitivním PCR (cPCR) je templátová i standardní (kompetitivní) DNA amplifikována v jedné zkumavce a díky mírně odlišné sekvenci nebo délce jsou potom odděleny gelovou elektroforézou. Množství templátové DNA je pak odhadnuto ze standardní křivky kompetitivní DNA.
Konečně real-time PCR měří DNA koncentraci průběžně během amplifikace monitorováním fluorescence. Jedná se v principu o klasickou PCR reakci, ve které je však množství PCR produktu (či jeho přírůstek) měřeno během každého cyklu reakce, v jednodušším případě nespecifickou reakcí syntetizované DNA s vhodným fluorescenčním barvivem (obvykle SYBR green), nebo specifickou (a dražší) reakcí syntetizované DNA s přítomnou DNA sondou. Množství vznikajícího PCR produktu je tedy možné kvantifikovat buď relativně porovnáním s jinými vzorky, nebo absolutně porovnáním s kalibrační přímkou za využití známého množství DNA. Tímto způsobem je možné určit počáteční koncentraci templátové DNA bez použití kompetitivní DNA. Amplifikační křivka se skládá ze tří částí: v první „rovinné“ části (tzv. background) je množství nasyntetizované DNA pod detekčním limitem metody/přístroje. Následuje exponenciální část, kde dochází k exponenciálnímu nárustu množství detekované DNA a poté je dosaženo plató, kdy fluorescenční signál zůstává konstantní vzhledem k nasycení systému (Obr. 3-8). Čím více je ve zkoumané templátové DNA kopií detekovaného úseku DNA, tím dříve (v ranějším cyklu reakce) dojde k exponenciální fázi (a posléze i nasycení reakce).
Obr. 3-8 Real time PCR
(převzato z http://www.generi-biotech.com/real-time-pcr-sondy-kvantitativni-real-time-pcr/)
Real-time PCR se dnes i vzhledem ke klesajícím cenám tohoto zařízení stává součástí mnoha environmentálních studií.
DNA microarray
Jednou z významných aplikací PCR kombinovanou s DNA-DNA hybridizací, která byla poprvé využita před zhruba 20 lety, jsou DNA čipy (DNA chips), nebo také DNA microarrays. Jedná se o soubor velkého množství mikroskopických DNA sond (až 250.000 na jedné destičce) uchycených na pevném nosiči (předchůdcem této metody je Southern hybridizace), ke kterým se za podmínek vysoké specificity hybridizují značené cDNA/RNA ze zkoumaného vzorku. Možnosti aplikace této techniky jsou všestranné, od určování identity bakteriálních druhů (Cho a Tiedje, 2001), přímé detekce 16S rRNA (a jim odpovídajících mikroorganismů) v půdním extraktu (Small et al., 2001), detekce genů v životním prostředí (Pathak et al., 2010) a změny jejich exprese (Mathioni et al., 2011) až po hodnocení mikrobiální diverzity environmentálních vzorků (Greene a Voordouw, 2003). K nesporným výhodám této metody patří fakt, že může současně analyzovat tisíce genových sekvencí. Zároveň je ale nutné brát v úvahu značnou cenu technického zázemí potřebného k této metodě i cenu tvorby takovýchto čipů, které by nebylo ekonomické připravovat k jednomu použití/experimentu, ale předurčuje tuto metodu k opakované aplikaci při studiu nejrůznějších komplexních problémů/komunit.
Klonování, sekvenování a fylogenetická analýza
K základním metodám určení diverzity mikrobiálního společenstva patří klonování PCR produktů (samozřejmě za předpokladu vytvoření dostatečně velké knihovny), sekvenování získaných klonů a následná fylogenetická analýza. K tomuto účelu je možné využít buď gen pro 16/18S rRNA, případně jiný vhodný gen dle účelu studie. V případě použití univerzálních 16/18S rRNA primerů můžeme studovat diverzitu celého mikrobiálního společenstva, je také ale možné se soustředit na menší skupinu vhodným výběrem specifičtějších primerů. Podobné rozlišení je teoreticky možné i v případě primerů pro jednotlivé funkční geny. Základním předpokladem je zde dostatečná databáze sekvencí pro zkoumaný gen umožňující navržení vhodných primerů a v neposlední řadě i charakteristika vlastního genu, který by měl obsahovat části konzervativní pro vytvoření primeru a variabilní umožňující vlastní studium diverzity tak, jak je tomu u genu pro 16/18S rRNA. Po úspěšném vytvoření primerů následuje optimalizace PCR reakce a za předpokladu jejího úspěchu pak klonování vlastních PCR produktů a izolace klonů.
Před vlastním sekvenováním jednotlivých klonů je z ekonomických důvodů vhodné provést screening získaných klonů (například s použitím některé z výše uvedených metod jako například DGGE, T-RFLP a podobně), aby bylo možné sekvenovat vždy jen jednoho představitele z každé vytvořené skupiny představující potenciálně identické klony (představované například proužkem ve stejné poloze na DGGE gelu). Tento přístup však v sobě skrývá úskalí, že budou z analýzy vyloučeny klony jen zdánlivě identické (klony vytvářející identické proužky/bandy na DGGE gelu nemusí být – jak již bylo uvedeno výše – vždy identické) a tímto způsobem dojde k podhodnocení skutečné diverzity. Tento problém by mohlo v brzké budoucnosti odstranit snižování ceny sekvenování umožňující sekvenaci opravdu všech získaných klonů, tak jak se v minulosti již často dělo u mnohých zahraničních studií, kdy především výzkumné skupiny ze Spojených států, vzhledem k zanedbatelné ceně sekvenování ve velkých výzkumných centrech, zaplavily databáze sekvencemi z nejrůznějších – především mořských – habitatů, což mělo za následek jednak zkreslení výsledků studií z odlišných, především terestriálních habitatů a v neposlední řadě ztížení konkurence pro publikování výsledků u skupin nedisponujících těmito možnostmi.
Po úspěšném sekvenování je možné srovnat získané sekvence s vhodnou databází, jako je například BLAST (http://blast.ncbi.nlm.nih.gov/), The Basic Local Alignment Search Tool. Tento program najde podobnost vložené sekvence (ať už na úrovni nukleotidů, nebo proteinů) se sekvencí v databázi, spočítá statistický význam této shody a může naznačit funkční a evoluční vztah mezi sekvencemi. Dále je možné provést srovnání všech získaných sekvencí za účelem vytvoření dendrogramu. K tomuto účelu je možné požít mnoho programů. Jedním z nejjednodušších je například ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/), nebo jeho novější verze Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/). K sofistikovanějším programům patří například Phylip (the PHYLogeny Inference Package). Jedná se o soubor programů k určování fylogenetických vztahů a tvorbě evolučních dendrogramů. Výsledné dendrogramy je možné zobrazit v některém z jednodušších programů, jako je například TreeView (http://taxonomy.zoology.gla.ac.uk/rod/treeview.html).
ÚEB Biol, Přírodovědecká fakulta, Masarykova univerzita |
Návrat na úvodní stránku webu, přístupnost |
| Servisní středisko pro e-learning na MU
| Fakulta informatiky Masarykovy univerzity, 2014
Centrum interaktivních a multimediálních studijních opor pro inovaci výuky a efektivní učení | CZ.1.07/2.2.00/28.0041
