Anotace
Druh Treponema pallidum zahrnuje obligátní lidské patogeny, jež
mají podobnou velikost genomu a vysokou sekvenční homologii.
Poddruh pallidum (TPA) je původcem sexuálně přenosné syfilis,
poddruh pertenue (TPE) způsobuje nevenerické tropické
onemocnění yaws a poddruh endemicum (TEN) způsobuje
nevenerickou endemickou syfilis. Opičí izolát Fribourg-Blanc,
který řadíme k TPE kmenům, způsobuje onemocnění podobné yaws u
paviánů. Mezi další blízce příbuzná treponemata patří T.
paraluiscuniculi a T. paraluisleporis, původce králičí a zaječí
venerické syfilis. Všechna výše zmíněná treponemata nelze
kultivovat v podmínkách in vitro a jsou považována za
(nekultivovatelná) patogenní treponemata. Ačkoliv jednotlivá
patogenní treponemata nelze od sebe morfologicky ani
sérologicky rozeznat, klinická manifestace jednotlivých
onemocnění je značně odlišná. Metoda whole genome
fingerprinting (WGF) potvrdila více než 99,5% sekvenční
identitu mezi poddruhy TPA a TPE. Žádné rozsáhlé inzerce,
delece či chromozomové přestavby nebyly touto metodou objeveny.
Lze tedy předpokládat, že rozdílná patogenita je způsobena
pouze malými genetickými změnami. Aby bylo možné identifikovat
tyto malé genetické změny mezi TPE a dalšími poddruhy, tři
zástupci poddruhu TPE (Samoa D, CDC-2 a Gauthier) byly
podrobeny vysoce kvalitní celogenomové sekvenaci. Výsledné
genomové sekvence byly získány s použitím alespoň dvou
sekvenačních technik „nové generace“ (next-generation
sequencing), technikami komparativního genomového sekvencování,
454 pyrosekvencování nebo sekvencování metodou Illumina.
Paralogní oblasti, nejednoznačně určené nukleotidy a mezery
mezi kontigy byly sekvencovány dideoxyterminátorovou metodou.
Výsledné celogenomové sekvence byly ověřeny metodou WGF.
Experimentálně získaný restrikční profil byl porovnán s
restrikčním profilem získaným analýzou výsledné sekvence in
silico. Tento postup neodhalil žádný rozdíl. Proto je možné
předpokládat, že frekvence sekvenační chyby je nanejvýše 10-4.
Genom kmene TPE Samoa D, který obsahuje 1139330 bp, byl
sekvencován všemi třemi sekvenačními metodami nové generace,
kdežto kmeny CDC-2 (1139744 bp) a Gauthier (1139441 bp) byly
sekvencovány metodou 454 a Illumina. Aby bylo možné určit
kompletní sekvenci kmene Gauthier, musela býti vyvinuta metoda
Pooled Segment Genomic Sequencing. Chromozomální DNA kmene
Gauthier byla amplifikována v překrývajících se oblastech.
Vzniklé PCR produkty byly ekvimolárně smíchány a sekvencovány.
Genom kmene TPE Samoa D byl anotován de novo za použití
softwarů Glimmer3, FgenesB a GeneMark. Pro geny kódující
proteiny s nezmámou funkcí byla stanovena minimální hranice 150
bp. Genome kmene Samoa D se sestává z 1125 genů, z nichž 54 je
nepřekládáno (tyto geny kódují tRNA, rRNA a nekódující RNA).
Kódující oblasti zaujímají 95,36 % délky genomu. Celkově byly
anotovány 3 pseudogeny, 648 genů kódujících proteiny se známou
funkcí, 141 genů kódujících konzervovavé hypotetické proteiny o
neznámé funkci, 129 genů kódujících treponemální konzervovavé
hypotetické proteiny a 147 genů kódujících hypotetické
proteiny. Anotace kmene Samoa D byla použita jako předloha pro
anotaci kmenů TPE CDC-2, Gauthier a TPA DAL-1. U obou dalších
TPE kmenů byl zachován stejný počet anotovaných genů. Odlišný
počet nukleotidů v homopolymerních repeticích způsobil v genomu
CDC-2 posunovou mutaci ve 2 genech, které byly reanotovány. V
genomu Gauthier bylo reanotováno celkem 10 genů, včetně 9 genů
s mutací způsobující posun čtecího rámce (6 z nich se nacházelo
v oblasti homopolymerních repetic) a mutaci ve stop kodonu.
Celkem 16 genů bylo reanotováno u kmene TPA DAL-1, včetně 14
genů s mutací způsobující posun čtecího rámce (sedm z nich se
nacházelo v oblasti homopolymerních repetic), 2 genů se shluky
mnohočetných změn, 2 genových lokusů anotovaných jako 4 geny
(oba lokusy byly anotovány jako 2 geny) a 5 genů, které byly
zkráceny či prodlouženy v závislosti na manuální anotaci.
Nejvyšší vnitrokmenová a mezikmenová variabilita mezi zástupci
poddruhu TPE byla pozorována v oblasti tpr (Treponema pallidum
repeat) genů, jež jsou považovány za geny kódující virulenční
faktory. Největší vnitrokmenová heterogenita byla prokázána v
genu tprK (TPE_0897), naopak gen tprD (TPE_0131) vykazoval
největší variabilitu mezi kmeny. Vysoká mezikmenová i
vnitrokmenová variabilita byla sledována nejenom uvnitř tpr
genů, ale také v několika oblastech s variabilním počtem
nukleotidů uvnitř homopolymerní repetice. Nalelzené indely v
těchto oblastech mohou mít za následek posunovou mutaci či
rozdílnou regulaci genové exprese. Pouze v kmeni Gauthier byly
objeveny další mutace, které způsobují posun čtecího rámce u
celkově tří genů. Gen TPEGAU_0856a kóduje hypotetický protein a
geny TPEGAU_0629 a TPEGAU_0858 kódují treponemální konzervované
hypotetické proteiny. U všech vyšetřených TPE kmenů byl popsán
rozdílný počet tandemových repetic, které nezpůsobují posun
čtecího rámce. Počet 60bp tandemových repetic genu arp
(TPE_0433 kódujícího acidic repeat protein) kolísá mezi 4 a 12,
počet 24bp tandemových repetic genu TPE_0470 (konzervovaný
hypotetický protein) se nachází v rozmezí 12 a 37 a počet 9bp
tandemových repetic genu TPE_0967 (treponemální konzervovaný
hypotetický protein) alternuje mezi dvěmi a čtyřmi repeticemi.
Dva další indely, které nezapříčinily posun čtecího rámce, byly
identifikovány v genech TPE_0067 (protein, který se účastní
buněčného dělení) a TPE_0136 (protein vnější membrány). Oba
deletované úseky byly ohraničeny přímými repeticemi. Navíc gen
TPE_0136 nese další jednonukleotidové záměny. V rámci poddruhu
TPE byly nalezeny tři geny pod pozitivním selekčním tlakem.
Jsou to: tp92 (TPE_0326, protein vnější membrány, povrchový
antigen Tp92), gen mcp2 (TPE_0488, protein, který se účastní
chemotaxe) a TPE_0548 (treponemální konzervovaný hypotetický
membránový protein). Je tedy možné, že tyto geny kódují
virulenční faktory poddruhu TPE.
Některé z identifikovaných změn mezi TPE kmeny se mohou stát
vhodnými kandidáty pro molekulární diagnostiku a
epidemiologickou typizaci. Operony s geny kódující ribozomální
RNA (rrn) byly vyšetřeny ve 20 kmenech nekultivovatelných
patogenních treponemat, včetně 11 kmenů poddruhu TPA, 5 kmenů
poddruhu TPE, 2 kmenů poddruhu TEN, opičího izobátu
Fribourg-Blamc a králičího zástupce druhu T. paraluiscuniculi.
Každý vyšetřený kmen obsahoval 2 rrn operony, které se lišily v
oblasti 16S-23S ribozomálního intergenového mezerníku. Tento
mezerník obsahuje jeden gen kódující tRNA, a to buď gen pro
alanin-tRNA nebo gen kódující izoleucin-tRNA. Oba geny pro tRNA
jsou vždy přítomny v genomu, mění se tedy jejich lokalizace.
Pokud pomineme geny kódující odlišné tRNA produkty, pouze jedna
delece předcházející genu pro 16S rRNA a dalších 17
heterologních nukleotidů byly nalezeny v rrn operonech
treponemálních kmenů. Struktura konzervovaných a heterologních
oblastí rrn operonu odpovídala klasifikaci treponemálních
kmenů. Nicméně odlišná lokalizace genů pro tRNA (vzorec Ile/Ala
nebo Ala/Ile) se zdá býti náhodná navzdory druhové a poddruhové
klasifikaci, času a místa původu izolátů. Oblast 16S-23S
ribozomálního intergenového mezerníku byla dále ještě vyšetřena
u 30 klinických izolátů (pocházejících z České republiky),
které náležely do pěti odlišných genotypů. Všechny klinické
izoláty obsahovaly stejný vzorec (Ile/Ala), kdy gen pro
izoleucin-tRNA byl lokalizován v rrn1 a gen pro alalnin-tRNA
byl umístěn v rrn2 operonu. Reciproká translokace rrn operonů
je pravděpodobně zajištěna rekombinačním mechanizmem, který je
podobný mechanizmu dráhy recBCD. Nejmenší sada (19 klonů) BAC
knihovny kmene TPA Nichols byla podrobena testům zkoumajícím
rozličné fenotypové projevy. Celkově bylo provedeno 1342
paralelních testů; 190 substrátů bylo testováno jako jediných
zdroj uhlíku, 192 testů sledovalo citlivost bakterií k různému
pH a osmotického prostředí a 960 testů prověřovalo rezistenci k
antibiotikům. Tři klony BAC knihovny vykazovaly zvýšenou
rezistenci. Klon DSTP001 byl více rezistentní k lincomycinu a
cefalosporinům, klon DSTP334 vykazoval zvýšenou rezistenci k
antibiotikům cinoxacin a 2,4-diamino-6,7-diisopropyl-pteridin,
klon DSTP094 byl více rezistentní k antibiotikům guanazol a
D,L-serin hydroxamát. Konkrétní treponemální geny, které
zvyšují odolnost hostitelské Escherichia coli k výše zmíněným
antibiotikům, je nutné určit. …víceméně
Abstract
The species Treponema pallidum includes several obligate human
pathogens with similar genome sizes and a high degree of
sequence similarity. The subspecies pallidum (TPA) is an agent
of sexually transmitted syphilis, the subspecies pertenue (TPE)
causes non-venereal tropical disease yaws, and the subspecies
endemicum (TEN) causes non-venereal endemic syphilis. In
addition to human pathogens, the simian Fribourg-Blanc strain,
clustering within the TPE group, causes a yaws-like disease in
baboons. Other closely related treponemes include T.
paraluiscuniculi and T. paraluisleporis, the agents of rabbit
and hare venereal disease, respectively. All these organisms
cannot be continuously cultivated in vitro and they are
considered as non-cultivable pathogenic treponemes. The
diseases caused by these treponemes are quite distinct,
although the pathogens are microscopically and serologically
indistinguishable, being >99.5% identical based on the whole
genome fingerprinting (WGF) results. These results showed that
no major gene deletions, insertions or rearrangement are
present among pathogenic treponemes. Relatively subtle genetic
differences are thus responsible for different pathogenicity
and host range. To precisely define genetic differences between
TPE and other subspecies, high quality whole genome sequences
of three TPE strains (Samoa D, CDC-2, Gauthier) were determined
by a combination of several next-generation sequencing
techniques, including Comparative Genome Sequencing, 454
pyrosequencing and Illumina techniques. Paralogous regions,
ambiguous bases and gaps were resolved using dideoxy-terminator
sequencing. To verify the final genome assemblies, the WGF was
compared to the in silico restriction enzyme analysis of each
sequenced TPE genome. No discrepancies between in silico and
experimental restriction site analyses of TPE genomes were
found, thus estimating a sequencing error rate to be lower than
10-4.
The TPE Samoa D genome, comprising 1,139,330 bp, was sequenced
by all three next-generation sequencing techniques mentioned
above, while CDC-2 (1,139,744 bp) and Gauthier (1,139,441 bp)
genomes were determined by 454 and Illumina approaches. To
determine the Gauthier genome sequence, a pooled segment
genomic sequencing method needed to be developed. Briefly,
chromosomal DNA was amplified in overlapping segments and
equimolar PCR products were pooled and sequenced. The TPE Samoa
D genome was annotated de novo using Glimmer3, FgenesB and
GeneMark software. For genes with unknown function, a gene size
limit of 150 bp was applied. The genome consists of 1125 genes,
including 54 untranslated genes (coding for rRNAs, tRNAs and
other non-coding RNAs). The coding regions comprise 95.36% of
the genome length. In total, 3 pseudogenes, 648 genes encoding
proteins with predicted function, 141 genes encoding conserved
hypothetical proteins, 129 genes encoding treponemal conserved
hypothetical proteins, and 147 genes encoding hypothetical
proteins were annotated. The TPE Samoa D gene annotation was
used as a scaffold for the TPE CDC-2 and Gauthier, and TPA
DAL-1 annotation. The same number of genes was preserved in
both CDC-2 and Gauthier TPE strains. Two genes were
re-annotated in the CDC-2 genome due to frameshuft mutation
caused by different number of homopolymers within homopolymeric
strings. In total, 10 genes were re-annotated in the Gauthier
genome, 9 genes due to grameshift mutation (6 within
homopolymeric tract) and one gene with a mutation in the
stopcodon. In total, 16 genes needed to be re-annotated in the
TPA DAL-1 strain, including 14 genes carrying a frameshift
mutation (7 within homopolymeric tract) and 2 genes with a
cluster of multiple mutations. As a consequence, 4 Samoa D
orthologues were not annotated, 5 genes were considered
pseudogenes, 2 gene loci were annotated as 4 genes (each locus
was split into 2 genes) and 5 genes were truncated or elongated
based on manual prediction. The major intra- and inter-strain
variability among TPE strains was observed within tpr
(Treponema pallidum repeat) genes, genes coding for putative
virulence factors. Whereas tprK (TPE_0897) gene showed the
highest intra-strain heterogeneity, the tprD (TPE_0131) gene
showed the highest heterogeneity between strains. In addition
to high variability observed in tpr genes, different numbers of
nucleotides within several homopolymeric strings were revealed
between and within TPE strains. Indels within homopolymeric
strings can result in frameshift mutation or different gene
expression.
Additional frameshift mutations were observed only in the
Gauthier strain, affecting three genes coding for hypothetical
(TPEGAU_0856a) or treponemal conserved hypothetical proteins
(TPEGAU_0629, TPEGAU_0858). Among all examined TPE strains,
three genes with variable number of tandem repeats do not
disrupt an open reading frame. The number of 60-bp tandem
repeat in the arp gene (TPE_0433 encoding acidic repeat
protein) varies between 4 and 12, the number of 24-bp tandem
repeats in TPE_0470 (conserved hypothetical protein) is between
12 and 37, and the number of 9-bp tandem repeats in TPE_0967
(treponemal conserved hypothetical protein) alternates between
two and four.
Two other indels, preserving the open reading frames, were
identified in TPE_0067 (cell division protein) and TPE_0136
(outer membrane protein). Interestingly, the deleted loci were
terminated by direct repeats. Moreover, TPE_0136 harbors
additional single nucleotide mutations. Within TPE strains,
three genes were found to be under positive selection,
including tp92 (TPE_0326, outer membrane protein), mcp2
(TPE_0488, methyl-accepting chemotaxis protein) and TPE_0548
(treponemal conserved hypothetical membrane protein). These
genes likely encode virulence factors or antigens. The
identified changes that are specific to individual strains
represent suitable targets for molecular diagnostics and
epidemiologic typing. The rrn operons were examined in 20
strains of non-cultivable pathogenic treponemes, including 11
strains of TPA, 5 strains of TPE, 2 strains of TEN, a simian
Fribourg-Blanc and a rabbit T. paraluiscuniculi strain. Every
examined strain carries 2 rrn operons, each with a different
16S-23S ribosomal intergenic spacer, containing a gene encoding
either tRNA-Ala or tRNA-Ile. With the exception of genes coding
for tRNAs, only a deletion upstream of the 16S rDNA and
additional 17 heterogenous nucleotide positions were found
within the rrn operons among treponemal strains. The sequence
of the rrn operons reflects the classification, while different
rrn spacer patterns (Ile/Ala and Ala/Ile) appeared to be
randomly distributed across the species/subspecies
classification, time, and geographical source of the treponemal
strains. Moreover, 16S-23S ribosomal intergenic spacers were
determined for 30 clinical samples (of the Czech Republic
origin) belonging to 5 different genotypes. All clinical
samples showed the Ile/Ala pattern. The reciprocal
translocation of genes coding for tRNA is likely mediated by a
recBCD-like recombination pathway. A minimal set (19 clones) of
the BAC library of TPA Nichols DNA was tested under 1342
different phenotype conditions. Overall, 190 substrates were
tested as sole carbon sources and 192 pH and osmotic
sensitivity and 960 antibiotic resistance assays were
performed. An increased resistance to lincomycin and
cephalosporins was observed in the DSTP001 clone, resistance to
cinoxacin and 2,4-diamino-6,7-diisopropyl-pteridine in the
DSTP334 clone, and resistance to guanazole and D,L-serine
hydroxamate in the DSTP094 clone when compared to other clones
and the host E. coli strain. The treponemal genes causing the
higher resistance of these BAC clones need to be further
examined. …víceméně
 angličtina
angličtina
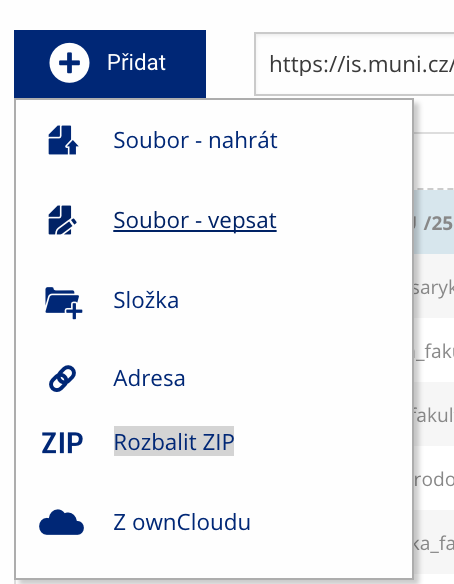 Soubor nebo složku lze nahrát pomocí tlačítka Přidat.
Soubor nebo složku lze nahrát pomocí tlačítka Přidat.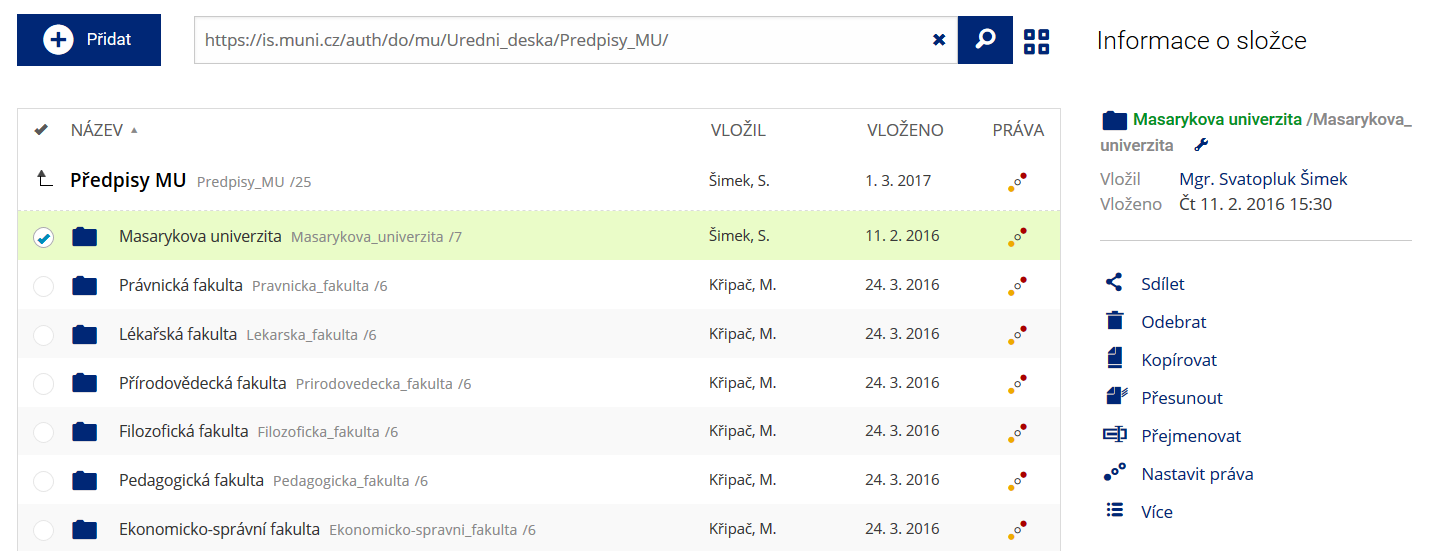 Podrobnosti lze zjistit označením příslušného řádku.
Podrobnosti lze zjistit označením příslušného řádku.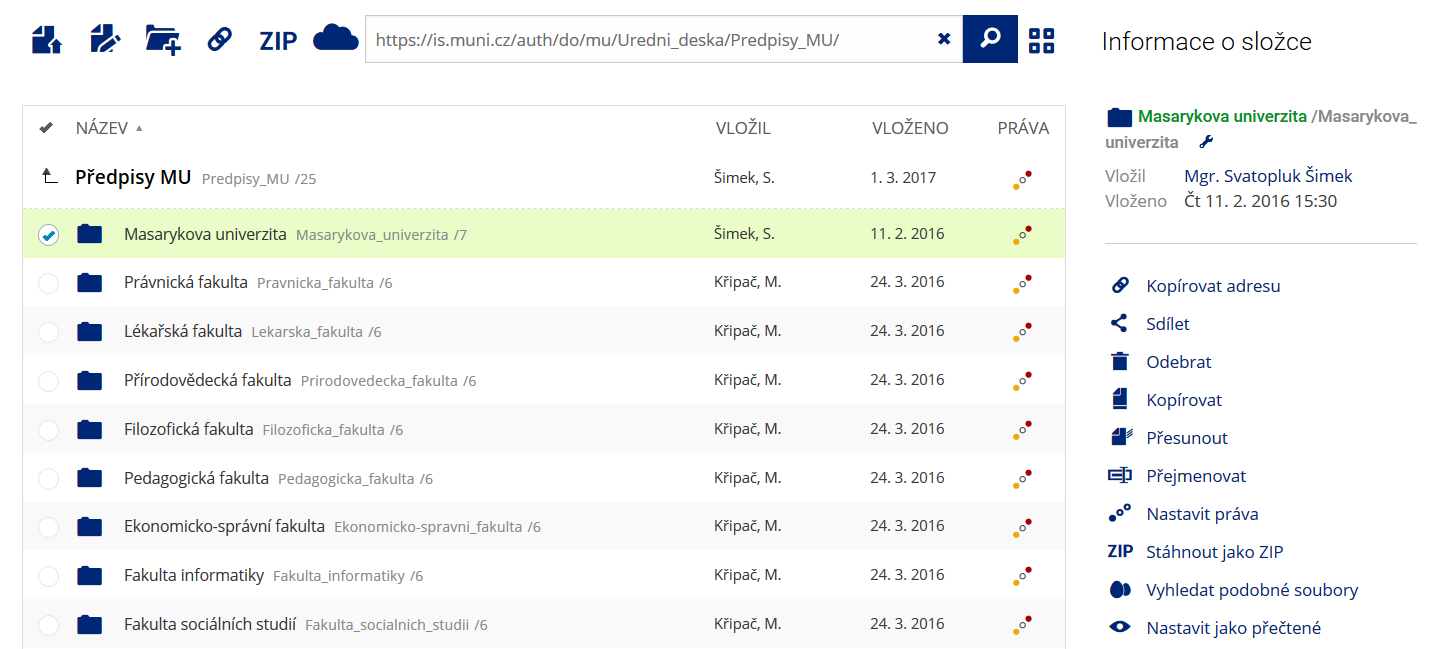 Pro častou práci je možné zvolit režim Více možností.
Pro častou práci je možné zvolit režim Více možností. Vyhledávaný výraz můžete zadat přímo do adresního řádku.
Vyhledávaný výraz můžete zadat přímo do adresního řádku. Pomocí funkce Nedávné je možné se rychle vrátit k právě prohlíženým souborům. Oblíbené soubory je také možné označit Hvězdičkou.
Pomocí funkce Nedávné je možné se rychle vrátit k právě prohlíženým souborům. Oblíbené soubory je také možné označit Hvězdičkou.