21 Základy termodynamiky
21.1 Termodynamika
Energetickou stránkou popisu soustav a jejich změn se zabývá obsáhlá fyzikální disciplína – termodynamika. Z široké oblasti obecné termodynamiky zajímá chemiky hlavně chemická termodynamika, která se zabývá především popisem chemických dějů z energetického hlediska.
21.1.1 Základní pojmy termodynamiky
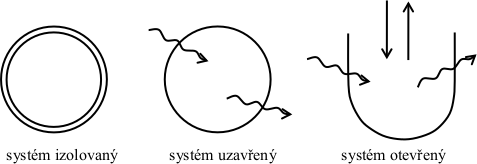
Termodynamická soustava (systém) je ta část prostoru, kterou sledujeme. Zbytek prostoru nazýváme okolí. Systém může být:
- izolovaný – tento systém nevyměňuje s okolím energii ani u něj nenastává látková výměna s okolím – např. obsah uzavřené Dewarovy nádoby (tzv. termoska);
- uzavřený – tento systém vyměňuje s okolím energii, ale není u něj látková výměna s okolím – např. obsah uzavřené konzervy;
- otevřený – tento systém vyměňuje s okolím energii i látky – např. živé organismy.
 … energie,
… energie, … látky
… látky
Část systému, která má stejné fyzikální i chemické vlastnosti v celém svém objemu, se nazývá fáze. Pojem „fáze“ není totožný s pojmem „skupenství“. Dá se říct, že skupenství je jedním z druhů fází, stejně jako např. různé krystalografické modifikace (Tab. 37).
| skupenství: | pevné | kapalné | plynné | 3 skupenství | ||
|---|---|---|---|---|---|---|
| fáze: | S (\(\ce{\alpha}\)) | S (\(\ce{\beta}\)) | S (amorfní) | S (kapalina) | S (plyn) | 5 fází |
Systémy můžeme klasifikovat podle počtu fází na:
- homogenní– systém je tvořen jednou fází;
např.:
vzduch – pouze plynná fáze,
roztok cukru ve vodě – pouze kapalná fáze,
bronz (slitina \(\ce{Sn + Cu}\)) – pouze jedna pevná fáze
- heterogenní– systém je tvořen nejméně dvěma fázemi;
např.:
olej smíchaný s vodou (2 kapalné fáze),
led ve vodě (kapalná a pevná fáze),
diamanty zahrabané v sazích (2 pevné fáze)
Stavové veličiny rozdělujeme na:
- intenzivní – jejich hodnota nezávisí na velikosti soustavy, např. koncentrace, teplota, hustota
- extenzivní – jejich hodnota závisí na velikosti soustavy, např. objem, hmotnost
Rozdíl mezi intenzivními a extenzivními veličinami si můžeme představit např. způsobem znázorněným na Obr. 21-2. Předpokládejme, že máme velkou kádinku plnou roztoku. Z kádinky část tohoto roztoku odlijeme do zkumavky. Ty vlastnosti, které bude mít roztok v obou nádobách stejné (koncentrace, hustota, teplota), nazýváme intenzivní. Ty vlastnosti, které se budou lišit (objem, hmotnost), nazýváme extenzivní.

Obr. 21-2: Znázornění vybraných intenzivních a extenzivních vlastností 14% vodného roztoku NaCl.
Intenzivní vlastnosti jsou zarámečkované.
Přechod systému z jednoho stavu do druhého se nazývá termodynamický děj a může být:
- vratný (reverzibilní) – děj lze kdykoli zastavit a systém stejnou cestou vrátit do původního stavu (např. sundat hrneček z poličky a zase jej tam vrátit)
- nevratný (ireverzibilní) – všechny ostatní děje (např. shodíme hrneček z poličky, rozbijeme jej, koupíme nový a do poličky „vrátíme“ nový)
Probíhá-li výměna látek i energie mezi systémem a okolím oběma směry stejnou rychlostí, je soustava v termodynamické rovnováze. Tu je nutné odlišovat od ustáleného stavu (viz kapitola 22).
Stavovou funkcí je například vnitřní energie souboru molekul (vnitřní energie se obvykle značí U). Skládá se z více příspěvků (Obr. 21-3).
\(\ce{U}\) vnitřní energie
\(\ce{E_{trans}}\) translační energie molekul
\(\ce{E_{el}}\) energie elektronů v orbitalech (související se základním či excitovaným stavem molekuly)
\(\ce{E_{rot}}\) rotační energie molekul
\(\ce{E_{vib}}\) vibrační energie molekul

Obr. 21-3: Příspěvky k pohybu molekul.
vlevo – translace (posun molekul v prostoru), uprostřed – rotace,
vpravo – vibrace (periodické změny vazebných délek nebo vazebných úhlů).
Je zřejmé, že s ohledem na počet molekul ve vzorku běžné velikosti a na statistické rozložení rychlostí pohybu molekul i jejich částí není možné celkovou hodnotu vnitřní energie vypočítat. Změřit ji také není možné, protože při každém měření se změní některá ze stavových veličin danou soustavu charakterizujících.
21.1.2 První věta termodynamická
První věta termodynamická je v podstatě zákon zachování energie aplikovaný na termodynamické děje.
Oddělíme-li z celkové energie soustavy její celkovou energii kinetickou a potenciální, zbude tzv. vnitřní energie soustavy \(\ce{U}\). Jedná se o stavovou funkci. Při přechodu ze stavu 1 do stavu 2 se změní vnitřní energie soustavy o \(\MR{\Delta U = U2 - U1}\).
Energie se mezi soustavami může vyměňovat dvěma způsoby: ve formě práce \(\ce{W}\) nebo ve formě tepla \(\ce{Q}\). Jednotky všech těchto tří veličin (\(\ce{U, W, Q}\)) jsou stejné, a to joule \(\ce{J}\).
Matematickým vyjádřením první věty termodynamické je vztah:
\(\mathrm{\Delta U = Q + W}\tag{21-1}\)Vzrůst vnitřní energie soustavy je roven součtu tepla a práce, které soustava při tomto ději přijala.
Je nutno odlišovat práci vykonanou soustavou \(\ce{\overline{W}}\) a práci vykonanou vnějšími silami \(\ce{W}\). Mezi oběma platí vztah:
\(\mathrm{W = - \overline{W}}\tag{21-2}\)Práce
Práci dělíme na:
- objemovou – objem soustavy se při daném ději mění, týká se zejména plynů (rozepínání, stlačování),
- neobjemovou – objem soustavy se nemění, jde o práci vykonanou např. elektrickými silami.
Velikost objemové práce
Obecně pro velikost objemové práce platí vztah:
| \(\mathrm{V_1}\) | počáteční objem soustavy (\(\mathrm{m^3}\)) |
| \(\mathrm{V_2}\) | konečný objem soustavy (\(\mathrm{m^3}\)) |
| \(\mathrm{p}\) | tlak (\(\mathrm{Pa}\)) |
| \(\mathrm{V}\) | objem (\(\mathrm{m^3}\)) |
Z tohoto vztahu a z významu určitého integrálu plyne, že objemová práce vykonaná soustavou \(\ce{\overline{W}}\) je číselně rovna plošnému obsahu obrazce pod grafem závislosti tlaku na objemu v tzv. \(\ce{p-V}\) diagramu:
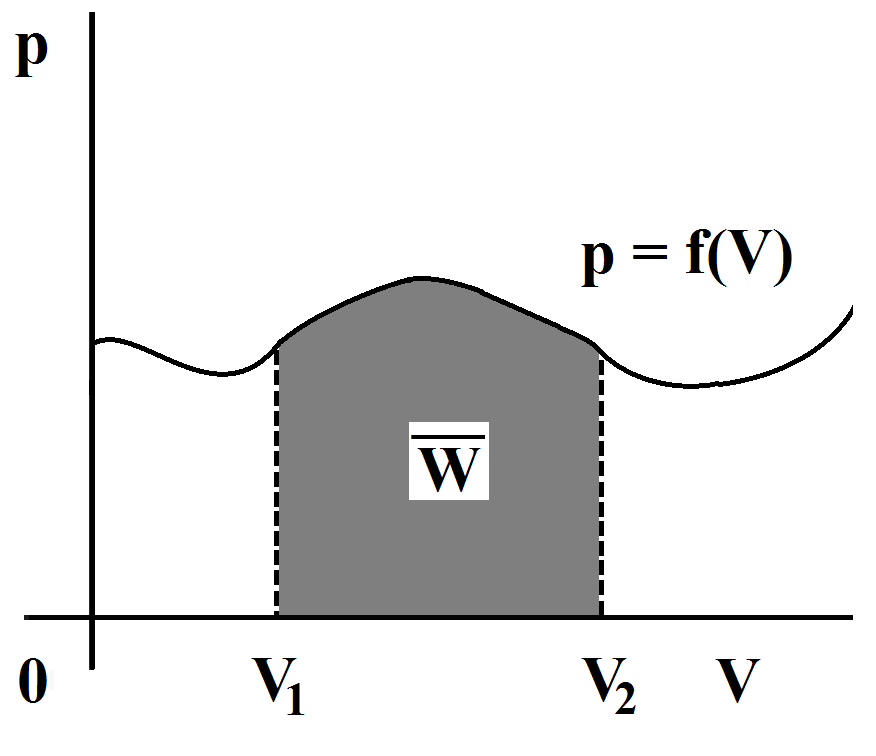
Následující tabulka ukazuje způsob výpočtu objemové práce \(\overline{W}\) při různých termodynamických dějích s plynem. Předpokládá se uzavřená soustava (tj. konstantní množství plynu, n = konst.).
Význam symbolů:
| \(V\) | objem, | \(V_1\) | počáteční objem, | \(V_2\) | konečný objem, |
| \(p\) | tlak, | \(T\) | termodynamická teplota, | \(n\) | látkové množství, |
| \(R\) | molární plynová konstanta. | ||||
| děj | p–V diagram | objemová práce \(\overline{W}\) vykonaná soustavou (výpočet) | výsledný vztah pro \(\overline{W}\) |
|---|---|---|---|
| izochorický V = konst. |
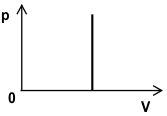 |
protože V = konst., odtud dV = 0 \(\Rightarrow \overline{W}=\displaystyle\int\limits_{V_1}^{V_2}pdV=0\) | \(\overline{W} = 0\) Při izochorickém ději se objemová práce nekoná. |
| izobarický p = konst. |
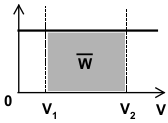 |
\(\overline{W}=\displaystyle\int\limits_{V_1}^{V_2}pdV = p\displaystyle\int\limits_{V_1}^{V_2}dV = p[V]_{V_1}^{V_2}=\\=p(V_2-V_1)\) (neboli plošný obsah vyšrafovaného obdélníka) | \(\overline{W} = p(V_2 − V_1)\) |
| izotermický T = konst. |
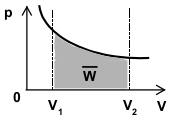 |
\(\overline{W}=\displaystyle\int\limits_{V_1}^{V_2}pdV=\lvert pV = nRT\Rightarrow p=\dfrac{nRT}{V}\rvert=\\=\displaystyle\int\limits_{V_1}^{V_2}\dfrac{nRT}{V}dV=\lvert n,R,T = \text{konst.}\rvert =\\=nRT\displaystyle\int\limits_{V_1}^{V_2}\dfrac{1}{V}dV=nRT[\ln V]_{V_1}^{V_2}=\\=nRT\left(\ln V_2 - \ln V_1\right) = nRT\ln\dfrac{V_2}{V_1}\) | \(\overline{W}=nRT\ln\dfrac{V_2}{V_1}\) |
21.2 Termochemie
Protože při chemických experimentech často pracujeme za konstantního (nebo téměř konstantního) tlaku, zavádíme pro teplo předané při izobarických dějích samostatnou fyzikální veličinu, kterou nazýváme enthalpie.
Reakční enthalpie \(\mathrm{\Delta H}\) je teplo, které reakční soustava přijme při reakci probíhající za konstantního tlaku, přičemž teplota výchozích látek a produktů zůstává stejná. Uskuteční-li se za těchto podmínek reakce v jednotkovém rozsahu (tj. zreagují-li v soustavě za konstantního tlaku taková látková množství jednotlivých složek, jaká udávají stechiometrické koeficienty v chemické rovnici), pak mluvíme o molární reakční entalpii.
Množství uvolněného či pohlceného tepla závisí nejen na druhu a množství reagujících látek, ale i na jejich skupenství a na způsobu, jakým reakce probíhá. Probíhá-li reakce za standardních podmínek (teplota \(\mathrm{25\, ^{\circ}C}\); tlak \(\mathrm{101,325\,kPa}\)), mluvíme o standardní reakční enthalpii a značíme ji \(\mathrm{\Delta H^{\circ}}\). Uskuteční-li se reakce za těchto podmínek v jednotkovém látkovém rozsahu, jde o standardní molární reakční entalpii.
Podle znaménka reakční enthalpie dělíme reakce na:
- exotermické – soustava teplo uvolňuje a předává ho okolí \(\mathrm{\left(\Delta H\lt 0\right)}\),
Příkladem exotermické reakce je hoření uhlíku:
\[\ce{C(s) + O2 (g) -> CO2 (g)}\qquad\mathrm{\Delta H = -398,8\,kJ\,mol^{-1}}\] - endotermické– soustava teplo pohlcuje \(\mathrm{\left(\Delta H\gt 0\right)}\), neboli přijímá od okolí.
Typickou endotermickou reakcí je redukce oxidu uhličitého uhlíkem:
\[\ce{CO2 (g) + C (s) -> 2 CO (g)}\qquad\mathrm{\Delta H = 173,6\,kJ\,mol^{-1}}\]
21.2.1 Termochemické zákony
První termochemický zákon


První termochemický zákon formulovali v roce 1780 A. Lavoisier a P.-S. Laplace.
Reakční teplo dané reakce a reakční teplo téže reakce, probíhající za stejných podmínek opačným směrem, je až na znaménko stejné:
\[\fbox{výchozí látky} \ce{<=>[\Delta H_1][\Delta H_2]} \fbox{produkty}\\ \mathrm{\Delta H_1= -\Delta H_2}\](horní řádek je Haber-Boschova syntéza, využívaná k průmyslové výrobě amoniaku):
\begin{array}{llll} \ce{1/2 N2 (g) + 3/2 H2 (g)-> NH3 (g)} & \quad & \ce{\Delta H_{298}^{\circ}} &\mathrm{= -46,0\,kJ\,mol^{-1}} \\ \ce{NH3 (g) -> 1/2 N2 (g) + 3/2 H2 (g)} & \quad & \ce{\Delta H_{298}^{\circ}} &\mathrm{= +46,0\,kJ\,mol^{-1}} \end{array}
Druhý termochemický zákon
Autorem druhého termochemického zákona je G. H. Hess, zákon zveřejnil v roce 1840.
Standardní enthalpie celkové reakce se rovná součtu standardních enthalpií jednotlivých reakcí, z nichž je možno danou reakci sestavit.

Odtud plyne, že celkový tepelný efekt chemické reakce je stejný pro všechny cesty od výchozích látek k produktům.

21.2.2 Využití termochemických zákonů
Oba termochemické zákony se používají k výpočtu reakčních tepel, která nelze získat přímým měřením (ať už z technických, nebo i časových či ekonomických důvodů).
Termochemické (enthalpické) děje
Existuje celá řada dějů doprovázených entalpickými změnami a s tím souvisejících různých entalpií, příp. energií dějů:
| enthalpie tání | vazebná / disociační energie |
| enthalpie tuhnutí | ionizační potenciál |
| enthalpie výparná | elektronová afinita |
| enthalpie kondenzační | |
| enthalpie sublimační | |
| enthalpie desublimační | |
| entalpie reakční |
Je zřejmé, že není možné v tabulkách uvádět reakční enthalpie všech myslitelných reakcí. Po dohodě bylo určeno, že z reakčních enthalpií budou tabelovány pouze tzv. standardní slučovací entalpie látek a standardní spalné enthalpie látek. Ostatní reakční enthalpie je možné ze slučovacích i spalných enthalpií vypočítat pomocí termochemických zákonů (viz výše).
Termochemické (enthalpické) cykly
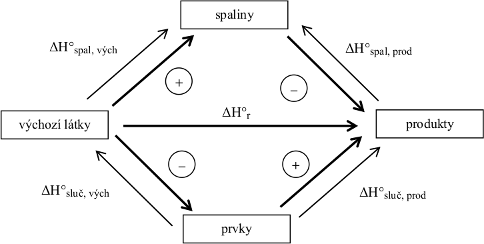
Tenká čára znázorňuje údaje, které lze najít v tabulkách, tlustá čára znázorňuje postup výpočtu a potřebnou změnu znaménka.
- Výpočet reakčního tepla obecné reakce ze standardních slučovacích entalpií látek
Podle 2. termochemického zákona lze pro termochemický výpočet nahradit původní reakci (tučná vodorovná šipka v Obr. 21-5) sledem jiných reakcí, pokud se shodují výchozí látky a produkty. Představíme si tedy, že původní reakci
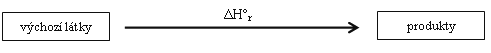
nahradíme sledem reakcí, což je v Obr. 21-5, dolní část, tučné šipky.
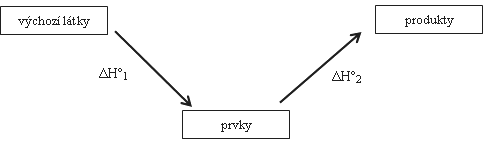
Enthalpii \(\mathrm{\Delta H_2}\) známe: je to \(\mathrm{\Delta H^\circ_\text{sluč, prod}}\) (Obr. 21-5 vpravo dole). Hodnotu \(\mathrm{\Delta H_1}\) neznáme, avšak známe enthalpii děje opačného, tj.
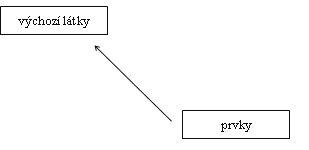 , což je \(\mathrm{\Delta H^\circ}\) sluč, vých (Obr. 21-5 vlevo dole).
, což je \(\mathrm{\Delta H^\circ}\) sluč, vých (Obr. 21-5 vlevo dole).
Pak zřejmě podle 1. termochemického zákona je
\[\mathrm{\Delta H_1 = -\Delta H^\circ_\text{sluč, vých}}\]Enthalpické změny v uvažovaném náhradním sledu reakcí pak jsou:
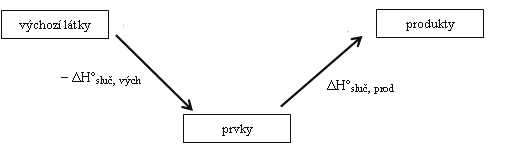
a podle druhého termochemického zákona je jejich součet roven celkové enthalpické změně původního uvažovaného děje:
\[\mathrm{\Delta H^\circ_r = - \Delta H^\circ_\text{sluč, vých} + \Delta H^\circ_\text{sluč, prod}}\] - Výpočet reakčního tepla obecné reakce ze standardních spalných enthalpií
Postupem analogickým bodu a. vyvodíme
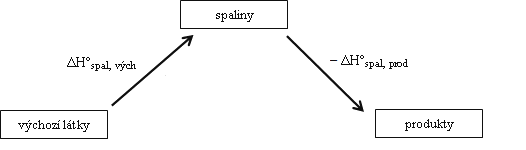
a odtud vztah
\[\mathrm{\Delta H^\circ r = \Delta H^\circ_\text{spal, vých} - \Delta H^\circ_{spal, prod}}\]
- Born-Haberův cyklus
Born-Haberův cyklus se používá při výpočtech týkajících se iontových sloučenin. Zde je ilustrován na příkladu výpočtu mřížkové energie LiF (s).
Uvažujme vznik sloučeniny LiF buď z iontů (horní část schématu), nebo slučováním prvků (dolní řádek schématu).
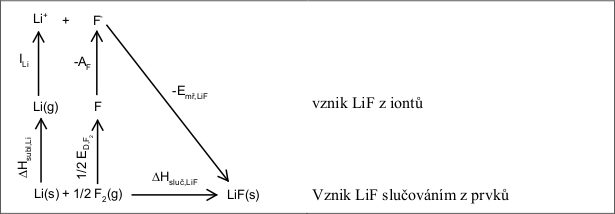 Obr. 21-6: Born-Haberův cyklus pro vznik LiF.
Obr. 21-6: Born-Haberův cyklus pro vznik LiF.\(\mathrm{\Delta H_{\text{sluč}, LiF}}\) standardní slučovací enthapie LiF \(\mathrm{\Delta H_{\text{subl}, Li}}\) sublimační enthalpie Li \(\mathrm{I_{Li}}\) ionizační potenciál Li \(\mathrm{E_{D, F_2}}\) disociační energie \(\mathrm{F_2}\) (\(\mathrm{=E_{vaz, F_2}}\) vazebná energie \(\mathrm{F_2}\) ) \(\mathrm{A_F}\) elektronová afinita F \(\mathrm{E_{\text{mř}, LiF}}\) mřížková energie LiF Energetické veličiny vystupující v tomto cyklu mají následující číselné hodnoty:
reakce tepelný efekt \(\ce{Li (s) + 1/2 F_2 (g) -> LiF (s)}\) \(\MR{\Delta H_{\text{sluč}, LiF} = -611,3\,kJ\,mol^{-1}}\) \(\ce{Li (s) \ce{->} Li (g)}\) \(\MR{\Delta H_{\text{subl}, Li} = 154,9\,kJ\,mol^{-1}}\) \(\ce{Li (g) \ce{->} Li^+ + e^-}\) \(\MR{I_{Li} = 519,2\,kJ\,mol^{-1}}\) \(\ce{1/2 F2 -> F}\) \(\ce{E_{D, F_2} = }\MR{150,8\,kJ\,mol^{-1} \Rightarrow }\ce{1/2 E_{D, F_2} = }\MR{75,4\,kJ\,mol^{-1} = }\ce{1/2} \MR{E_{\text{vaz}, F_2}}\) \(\ce{F + e^- -> F^-}\) \(\MR{A_F = 339,1\,kJ\,mol^{-1}}\) Pomocí 1. a 2. termochemického zákona lze pak odvodit, že pro děje znázorněné na Obr. 21-6 platí vztah:
\[\mathrm{\Delta H_{\text{sluč}, LiF} = \Delta H_{\text{subl}, Li} + I_{Li} + }\ce{1/2}\MR{ E_{D, F_2} - A_F - E_{\text{mř}, LiF}}\]Pro výpočet mřížkové energie rovnici upravíme a následně dosadíme číselné hodnoty:
\[\begin{array}{rcl} \mathrm{E_{\text{mř}, LiF}} & = & \mathrm{\Delta H_{\text{subl}, Li} + I_{Li} + }\ce{1/2}\MR{ E_{D, F_2} - A_F - \Delta H_{\text{sluč}, LiF}} \\ \mathrm{E_{\text{mř}, LiF}} & = & 154,9 + 519,2 + 75,4 - 339,1 - (- 611,3) \\ \mathrm{E_{\text{mř}, LiF}} & = & \mathrm{1\,021,7\,kJ\,mol^{-1}} \end{array}\] - Enthalpický cyklus kovalentních látek
Hodnota vazebné energie každé jedné kovalentní vazby závisí na tom, v jaké molekule a na jakém místě v molekule tato vazba je. Uvažujeme-li například molekulu \(\ce{CH4}\), můžeme z ní odtrhávat atomy \(\ce{H}\) jeden po druhém. Každý z dějů je jiný, proto mají tyto děje různé reakční entalpie. Postupná disociace:
\[\begin{array}{lcllclcl} \ce{CH4} & \ce{->} & \ce{CH3} & + & \ce{H} & \mathrm{\Delta H_1 = 422\,kJ\,mol^{-1}} & \text{(pro libovolný 1. odtržený atom H)} \\ \ce{CH3} & \ce{->} & \ce{CH2} & + & \ce{H} & \mathrm{\Delta H_2 = 364\,kJ\,mol^{-1}} & \text{(pro libovolný 2. odtržený atom H)} \\ \ce{CH2} & \ce{->} & \ce{CH} & + & \ce{H} & \mathrm{\Delta H_3 = 385\,kJ\,mol^{-1}} & \text{(pro libovolný 3. odtržený atom H)} \\ \ce{CH} & \ce{->} & \ce{C} & + & \ce{H} & \mathrm{\Delta H_4 = 335\,kJ\,mol^{-1}} & \end{array}\]Pro zjednodušení však budeme uvažovat, že všechny 4 vazby \(\ce{C-H}\) se rozštěpí současně (a určíme tím střední energii vazby, která se uvádí v tabulkách).
 Obr. 21-7: Enthalpický cyklus methanu.
Obr. 21-7: Enthalpický cyklus methanu.\(\mathrm{E_{\text{vaz}, \ce{C-H}}}\) vazebná energie \(\ce{C-H}\) \(\mathrm{\Delta H_{\text{sluč}, CH_4}}\) slučovací enthalpie \(\ce{CH4}\) \(\mathrm{E_{D, H_2}}\) disociační energie \(\ce{H2}\) (\(\MR{= E_{\text{vaz},} \ce{(H-H)}}\) vazebná energie \(\ce{H2}\)) \(\mathrm{\Delta H_{\text{subl}, C}}\) sublimační enthalpie \(\ce{C}\) Z Obr. 21-7 je zřejmé, že pro reakci
\[\ce{CH4 -> 4H (g) + C (g)}\]platí vztah
\[\mathrm{4 E_{\text{vaz}, \ce{C-H}} = - \Delta H_{\text{sluč}, CH_4} + 2 E_{D , H_2} + \Delta H_{\text{subl}, C}}\]Energetické veličiny vystupující v tomto cyklu mají následující hodnoty:
reakce tepelný efekt \(\ce{CH4 -> 2 H2 (g) + C (s)}\) \(\mathrm{\Delta H = - \Delta H_{\text{sluč}, CH_4} = - (- 74,94)\,kJ\,mol^{-1}}\) \(\ce{H2 (g) -> 2 H (g)}\) \(\mathrm{E_{D, H_2} = 436,26\,kJ\,mol^{-1} = E_{\text{vaz}, (\ce{H-H})}}\) \(\ce{C (s) -> C (g)}\) \(\mathrm{\Delta H_{\text{subl}, C} = 717,2\,kJ\,mol^{-1}}\) Dosadíme číselně a vypočteme vazebnou energii:
\[\begin{array}{rcl} \mathrm{4 E_{vaz, \ce{C-H}}} & = & \mathrm{- (- 74,94) + 2 · 436,26 + 717,2} \\ \mathrm{4 E_{vaz, \ce{C-H}}} & = & \mathrm{1\,664,7\,kJ\,mol^{-1}} \\ \mathrm{E_{vaz, \ce{C-H}}} & = & \dfrac{1\,664,7}{4}=\mathrm{416,2\,kJ\,mol^{-1}} \end{array}\]
21.2.3 Odhad směru spontánního průběhu termodynamických dějů
Při odhadu směru spontánního průběhu děje se používá tzv. Gibbsova energie. Její hodnota souvisí s enthalpií, entropií a termodynamickou teplotou vztahem:
| \(\mathrm{\Delta G}\) | přírůstek Gibbsovy energie |
| \(\mathrm{\Delta H}\) | přírůstek enthalpie |
| \(\mathrm{T}\) | termodynamická teplota |
| \(\mathrm{\Delta S}\) | přírůstek entropie |
Entropie vyjadřuje míru neuspořádanosti soustavy. Systém o více částicích má za jinak stejných podmínek větší entropii. Entropie je definována vztahem:
| \(\mathrm{\Delta S}\) | přírůstek entropie |
| \(\mathrm{Q}\) | teplo přijaté soustavou při izotermickém ději |
| \(\mathrm{T}\) | teplota, při níž soustava teplo přijímá |
21.2.4 Odhad uskutečnitelnosti reakce
Chceme-li zjistit, zda uvažovaný děj může nastat, stačí vypočítat jeho ΔG. Mohou nastat tři možnosti:
| \(\mathrm{\Delta G \lt 0}\) | uvažovaný děj nastane |
| \(\mathrm{\Delta G \gt 0}\) | uvažovaný děj nenastane, proběhne děj opačný |
| \(\mathrm{\Delta G = 0}\) | nebudou probíhat pozorovatelné změny žádným směrem (stav rovnováhy) |
21.2.5 Vliv teploty na směr reakce
Tyto úkoly budeme řešit pomocí hodnoty změny Gibbsovy energie \(\mathrm{\Delta G}\).
Odhadněte, jakým směrem bude probíhat reakce
\[\ce{\it NO + 1/2O2 <=> NO2\quad\Delta H\lt 0}\]
Pro uvažovaný děj evidentně platí \(\mathrm{\Delta S \lt 0}\) (entropie, jakožto neuspořádanost soustavy, je v tomto případě větší na levé straně rovnice, protože je tam větší počet částic). Řešíme pomocí vztahu
\[\mathrm{\Delta G=\Delta H - T\Delta S\quad (a)}\]ad a) Nízká teplota:
Součin \(\mathrm{T\Delta S}\) bude malé číslo a na pravé straně rovnice (a) převládne \(\mathrm{\Delta H \lt 0}\). Proto bude \(\mathrm{\Delta G \lt 0}\) a reakce bude probíhat zleva doprava ⇒ stabilní bude \(\ce{NO2}\).
ad b) Vysoká teplota:
Součin \(\mathrm{T\Delta S}\) bude velké číslo, proto bude součin \(\mathrm{T\Delta S}\) mít velkou zápornou hodnotu. Na pravé straně vztahu (a) převládne člen – \(\mathrm{T\Delta S}\) (i se z áporným znaménkem má kladnou hodnotu). Proto bude \(\mathrm{\Delta G \gt 0}\) a reakce bude probíhat zprava doleva ⇒ stabilní bude \(\ce{NO}\) a \(\ce{O2}\).
Poznámka:
Směr reakce v tomto případě bylo možno odhadnout i bez výpočtu, neboť obecně platí, že zvýšením teploty jsou podporovány rozkladné (disociační) děje.